

法令・告示・通達
底質調査方法の改定について
環水管127号
(環境庁水質保全局長から都道府県知事・政令市長あて)
標記調査方法(昭和50年10月28日付け環水管第120号本職通知「底質調査方法について」の別添)の全部を別添のとおり改定する。
今回の改定の主な内容としては、JIS K 0102(工場排水試験方法)に整合する方向で新しい技術を取入れたほか、近年、閉鎖性水域の富栄養化の原因物質として重視されている全窒素及び全りん、分析方法が著しく改良されたシアン化合物等を新たに追加した。
なお、改定された「底質調査方法」についても、通常の底質調査における分析方法等を定めたもので、特殊な条件のもとで、これによることが著しく不適当と認められる場合には、これの骨子に沿って必要な変更を行っても差し支えない。
また、この改定に伴い、底質の暫定除去基準(昭和50年10月28日付け環水管第119号本職通知)の一部を下記のとおり改定する。
おって、関係者に対して、この趣旨の周知徹底を図るとともに、今後とも底質調査及び底質改善対策の一層の推進を図られたい。
記
2 底質の分析方法等中「「底質調査方法」(昭和50年10月28日付け環水管第120号。以下「底質調査方法」という。)」を「「底質調査方法」(昭和63年9月8日付け環水管第127号。以下「底質調査方法」という。)」に改める。
〔別添)底質調査方法
昭和63年9月
環境庁水質保全局
目次
底質調査方法
- Ⅰ 採泥
- Ⅱ 分析方法
- 1 結果の表示
- 2 分析試料の調製
- 3 乾燥減量
- 4 強熱減量
- 5 水銀
- 5.1 総水銀
- 5.2 アルキル水銀化合物
- 6 カドミウム
- 7 鉛
- 8 鋼
- 9 亜鉛
- 10 鉄
- 11 マンガン
- 12 クロム
- 12.1 総クロム
- 12.2 酸溶出クロム
- 12.3 六価クロム
- 13 ひ素
- 14 シアン化合物
- 15 ポリ塩素化ビフェニル(PCB)
- 16 ヘキサクロロシクロヘキサン(HCH、旧称BHC)
- 17 硫化物
- 18 全窒素(ケルダール窒素)
- 19 全りん
- 20 過マンガン酸カリウムによる酸素消費量〔CODsed〕
- Ⅲ 溶出試験
- 1 溶出率の算定法
- 2 水銀
- 2.1 総水銀
- 2.2 アルキル水銀化合物
- 3 カドミウム
- 4 鉛
- 5 ひ素
- 6 ヘキサクロロシクロヘキサン(HCH、旧称BHC)
底質調査方法
Ⅰ 採泥
1 採泥時期
底質中に含まれる物質が、水利用に悪影響を及ぼす時期を含めることを原則とし、当該水域につき水質調査の実施が予定されている場合は、水質調査の実施時期に合わせることが望ましい。
2 採泥地点
2.1概況調査
海域、湖沼においては、調査対象水域の規模及び予想される汚染の程度に応じて均等に2~6kmメッシュで採泥地点を設けるものとする。主要な排水口周辺水域等においては、地点を増加する。
河川においては、原則として、主要な排水口の直下50m下流及び流下方向1kmごとの汚泥の堆積しやすい地点とする。水域の状況等により適宜地点を増加する。
2.2 精密調査
海域、湖沼においては調査対象水域に200~300mメッシュで採泥地点を設定するものとし、河口部等の堆積汚泥の分布状況が変化しやすい場所等においては必要に応じて地点を増加するものとする。
河川及び水路においては、幅の広いときにあっては50mメッシュで、幅の狭いときにあっては流下方向50mごとに汚泥の堆積しやすい場所を採泥地点とし、水域の状況等により適宜地点を増加する。
3 採泥方法
2の各採泥地点において、エクマンパージ型採泥器又はこれに準ずる採泥器によって3回以上底質を採取し、それらを混合して採泥試料とする。
ただし、概況調査の場合は、SK式採泥器又はこれに準ずるものを用いても差し支えない。探さ方向の調査が必要な場合には、柱状試料を採取することとし、この場合は、原則として底質表面から深さ1mごとの各位置において、その各10cm程度の泥を採取し、その位置の試料とする。なお採取は1回でも差し支えない。
SK式採泥器を用いる場合には、なるべく短時間で1回採取を行いそのものを試料とする。
4 採泥時に実施すべき事項
採泥日時、採泥地点(図示すること)、採泥方法(使用した採泥器の型名)、底質の状態(堆積物、砂、泥などの別、色、臭気など)及びpHは直ちに観測測定し記録する。試料はできるだけ速やかに分析する。直ちに分析が行えない場合には、温度を低く保っておくこととする。なお、調査の目的に応じてその他の項目を適宜追加する。
5 採泥時の試料の調製
採泥試料を清浄なポリエチレン製のバット等(測定重金属等の物質の吸着、溶出等がない材質のものを使用する。)に移し、小石、貝殻、動植物片などの異物を除いた後、均等に混合し、その500~1,000gを清浄なポリエチレンびん、ポリエチレン袋等(測定重金属等の物質の吸着、溶出等がない材質のものを使用する。)に入れて、実験室に持ち帰るものとする。
Ⅱ 分析方法
1 結果の表示
原則として、3乾燥減量の操作を行って得られた乾燥試料当りの濃度(mg/kg又はg/kg)で、有効数字3桁まで表示する。
2 分析試料の調製
2.1 湿試料
(1)器具及び装置
- (a)遠心分離器
- (b)2mm目のふるい 金属成分の分析に供する場合には、ナイロンやサラン製など、測定成分の物質の吸着や溶出等がない材質のものを使用する。
(2)操作
- (a)Ⅰ採泥5の試料を2mm目のふるいに通し、その適量を分取し、3000rpmで20分間遠心分離する。
- (b)上澄み液を捨て、残留物を十分混和し湿試料とする。
2.2 風乾試料
(1)器具
- (a)風乾用皿 測定成分の物質の吸着や溶出等がない材質のものを使用する。
- (b)2mm目のふるい 金属成分の分析に供する場合には、ナイロンやサラン製など、測定成分の物質の吸着や溶出等がない材質のものを使用する。
(2)操作
- (a)2.1によって調製した湿試料の適量を清浄な風乾用皿にとり、均一に広げ、直射日光をさけ、室温で空気中の湿度と平衡になるまで乾燥(風乾)させる。
- (b)風乾した試料は、塊を清浄な乳ばち(ガラス又はめのう製)を用いて軽く押し潰してほぐし、2mm目のふるいを通し、これを風乾試料とする。
2.3 乾燥試料
(1)器具及び装置
- (a)乾燥器105~110℃に調節できるもの。
- (b)試料乾燥用皿ガラス又は陶磁器製の蒸発皿等(注1)を用いる。
注(1)試料を入れ105~110℃で加熱したとき、測定成分の吸着や溶出等がない材質のもので、2.1の湿試料10g以上を入れ乾燥したとき、乾燥にむらが生じないよう試料の厚さを約1cm以下になるように拡げて入れられる大ささのもの。
(2)操作
- (a)試料乾燥用皿に、2.1の湿試料から分析に必要な量を取り、厚さが1cm以下になるよう出来るだけ平らに拡げる。
- (b)105~110℃の乾燥器中で約2時間乾燥した後、デシケータ(注2)中で約40分間放冷する。乾燥により試料が現状に固まったときは、乳ばちなどを用いて軽く砕きほぐし、2mm目のふるいを通し、これを乾燥試料とする。乾燥試料は適当な容器(測定成分の汚染等のおそれのない材質のもの)に入れ密栓して保存する。
注(2)デシケータ中には、シリカゲル又は塩化カルシウムのいずれかの乾燥剤を用いる。
3 乾燥減量
(1)器具及び装置
- (a)乾燥器 2.3(1)(a)と同じ。
- (b)共栓はかりびん=105~110℃の乾燥器で加熱乾燥した後、デシケータ中で約40分間放冷し質量を0.01gのけたまで測定しておく。
(2)操作
- (a)2.1の湿試料から5g以上を共栓はかりびんに取り、厚さが1cm以下になるように拡げ、0.01gのけたまで質量を測定する。
- (b)105~110℃の乾燥器中で約2時間乾燥した後、デシケータ中で約40分間放冷し0.01gのけたまで質量を測定する。
- (c)次式により乾燥減量(%)を算出する。

注(1)共栓はかりびんに代えて磁器製のるつぼを用いた場合は、乾燥減量を測定した後、強熱減量の測定を行うことができる。この場合るつぼは、4(1×b)に準じて質量を測定しておく。このとき試料は厚さが1cm以下になるようにとる。
4 強熱減量
(1)器具及び装置
- (a)電気炉 600±25℃に調節できるもの。
- (b)るつぼ 磁器製のもの。600±25℃で約1時間強熱した後、デシケータ中で放冷し質量を1mgのけたまで測定しておく。
(2)操作
- (a)2.3(2)(b)で乾燥した乾燥試料5g以上を磁器製のるつぼに1mgのけたまではかり取る。
- (b)電気炉を用い600±25℃で約2時間強熱した後、デシケータ中で放冷し、質量を1mgのけたまで測定する。
- (c)次式により強熱減量(%)を算出する。

5 水銀
総水銀とアルキル水銀化合物に区分する。
5.1 総水銀(注1)
注(1)試験溶液の調製は、試料の性状により次の1~3から、適当なものを選んで行う。
- 1 硫酸-過マンガン酸カリウム還流分解法:還流冷却器分解フラスコを用い、硝酸と過マンガン酸カリウムにより前処理を行う方法で、試料中に有機物や硫化物などの多い試料に適用する。
- 2 硝酸-硫酸-過マンガン酸カリウム分解法:三角フラスコ又はケルダールフラスコを用い、硝酸、硫酸及び過マンガン酸カリウムにより温水浴中で分解処理を行う方法で、試料中の有機物等の分解が容易で、加熱操作中に加えた過マンガン酸カリウムの色が消えない試料に適用する。
- 3 硝酸-塩化ナトリウム分解法:ケルダールフラスコを用い、硝酸と塩化ナトリウムにより前処理を行う方法で、有機物等が少なく、試料中の水銀化合物が容易に分解でき、加熱操作中に水銀の損失が無い場合に適用する。
5.1.1硝酸-過マンガン酸カリウム還流分解法
(1)試薬
- (a)硝酸 水銀0.0001mg/l以下のもの。
- (b)硫酸(1+1)水銀0.001mg/l以下の硫酸を用いて調製する。
- (c)過マンガン酸カリウム溶液(3w/v%)過マンガン酸カリウム(注2)
15gを水に溶かしてガラスフィルターでろ過した後、水で500mlとする。硬質ガラスびんに保存する。 - (d)塩化ヒドロキシルアンモニウム溶液(20w/v%)塩化ヒドロキシルアンモニウム(塩酸ヒドロキシルアミン)20gを水に溶かして100mlにする。この溶液の水銀含有量は0.001mg/l以下であること。精製の必要がある場合は、この溶液を分液漏斗に移し、ジチゾン四塩化炭素溶液(0.005w/v%)少量を加えて振り混ぜ、放置後、四塩化炭素層を捨てる。この操作を四塩化炭素層が変色しなくなるまで繰り返す。水層を乾いたろ紙でろ過して四温化炭素の小粒を除く。
- (e)塩化すず(Ⅱ)溶液塩化すず(Ⅱ)二水和物10gに硫酸(1+20)60mlを加え、かき混ぜながら加熱して溶かす。放冷後、水を加えて100mlにする。この溶液の水銀含有量は0.001mg/l以下であること。精製の必要がある場合は、窒素を通気する。この溶液の保存期間は1週間以内とする。
- (f)水銀標準液(0.5mgHg/ml)塩化水銀(Ⅱ)0.339gを水に溶かす。全量フラスコ500mlに移し、硝酸(1+1)5mlを加えた後、水を標線まで加える。硬質ガラスびんに保存する。
- (g)水銀標準液(0.01mgHg/ml)水銀標準液(0.5mgHg/ml)10mlを全量フラスコ500mlにとり、硝酸(1+1)5mlを加え、水を標線まで加える。硬質ガラスびんに保存する。この溶液の保存期間は1か月以内とする。
- (h)水銀標準液(0.1μgHg/ml)水銀標準液(0.01mgHg/ml)10mlを全量フラスコ1000mlにとり、硝酸(1+1)10mlを加えた後、水を標線まで加える。使用時に調製する。
注(2)原子吸光分析用試薬など、水銀含有量の少ないものを用いる。
(2)器具及び装置
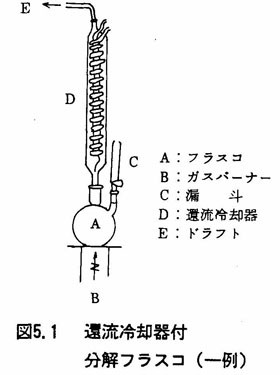
- (a)還流冷却器付分解フラスコ
(例を図5.1に示す)
- (b)原子吸光分析装置又は水銀用原子
吸光分析装置 - (c)水銀還元気化装置(注3)原子吸光分析
装置と併用する。 - (d)水銀中空陰極ランプ又は水銀ランプ
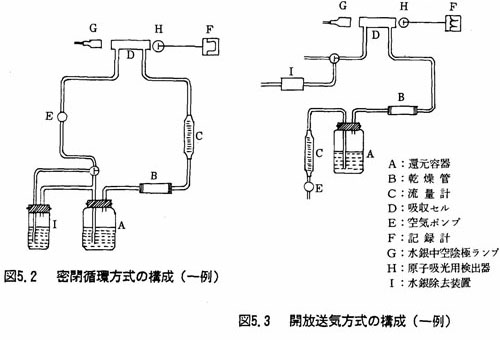
注(3)還元容器、吸収セル、空気ポンプ、流量計*、乾燥管及び連結管から横成される。図5.2(密閉循環方式)及び図5.3(開放送気方式)に構成を示す。
*流量計を用いないものもある。
なお、各構成部分の例は、次のとおりである。
還元容器 ガラスびん(又は三角フラスコ)300~350ml(250mlの位置に印を付けておく)
吸収セル 長さ100~300mm程度の石英ガラス製のもの又はガラス製、プラスチック製(水銀蒸発を吸着しないもの)で、両端に石英ガラス窓を付けたもの。
空気ポンプ 0.5~3l/minの送気能力をもつダイアフラムポンプ又は同じ性能をもつ空気ポンプ。水銀蒸気に接する部分が金属製の場合はコロジオンなどを塗布しておく。
流量計 0.5~5l/minの流量が測定できるもの。
乾燥管 乾燥塔又はU字管、粒状の過塩素酸マグネシウム、塩化カルシウムなどを充てんしておく。又はコールドトラップで代用してもよい。又は吸収セルの部分に小形電球を点灯するなどして吸収セル内の温度が周囲の温度よりも約10℃高くなるようにしておけば、乾燥管を用いなくてもよい。
連結管 軟質塩化ビニル管
(3)試験溶液の調製
- (a)2.1の湿試料約10gを0.1gのけたまではかり取り、これを還流冷却器付分解フラスコに入れ、硝酸(1+1)50mlを加え加熱し、穏やかに煮沸して有機物を分解する。
- (b)室温まで冷却して過マンガン酸カリウム溶液(3w/v%)20mlを加え、1時間加熱を続ける。
もしこの間に過マンガン酸カリウムの色が消える場合は、室温まで冷却した後過マンガン酸カリウム溶液(3w/v%)10mlを追加して、再び加熱する。 - (c)この操作を過マンガン酸カリウムの赤紫色が約10分間残るまで繰り返す(注4)。
- (d)液温を約40℃とし、尿素溶液(10w/v%)10mlを加え溶液を振り混ぜながら、塩化ヒドロキシルアンモニウム溶液(20w/v%)を滴加し、過剰の過マンガン酸カリウムを分解する。
- (e)これをガラス繊維又はガラス繊維ろ紙でろ過し、全量フラスコ200mlに入れ、水を標線まで加え、これを試験溶液とする。
- (f)出来るだけ速やかに、(4)操作を行う。
注(4)(e)の試験溶液について直ちに(4)の操作が行えない場合は、この状態で放冷し、保存する。
(4)操作
- (a)試験溶液の適量(Hgとして0.1~2μgを含む量)を還元容器に取り、これに硫酸(1+1)10mlと水を加えて約250mlとした後、通気回路を組み立てる。
- (b)手早く塩化すず(Ⅱ)溶液10mlを加え、あらかじめ設定した最適流量(注5)で空気ポンプを作動し、空気を循環(注6)させる。
- (c)波長253.7nmの指示値(7)読む。
- (d)バイパスコック(8)を回して、指示値が元に戻るまで通気を続ける。
- (e)空試験として(3)(a)~(f)のうち(a)の試料のはかり取りを除く操作、及び(a)~(d)の操作について試験溶液と同様に行って指示値(7)読み、試験溶液について得た指示値を補正する。
- (f)検量線から水銀の量を求め、分析試料中の水銀の濃度を算出する。
- (g)3乾燥減量で求めた分析試料の乾燥減量(%)で(f)の水銀濃度を補正し、乾燥試料当りの濃度(mgHg/kg)として結果を表示する。
検量線 水銀標準液(0.1μgHg/ml)1~20mlを還元容器に段階的にとり、硫酸(1+1)10mlと水を加えて約250mlとした後、通気回路を組み立て、(b)~(d)の操作を行う。
別に還元容器に硫酸(1+1)10mlをとり水を加えて250mlとした後、通気回路を組み立て、(b)~(d)の操作を行って、水銀標準液について得た指示値を補正し、水銀(Hg)の量と指示値との関係線を作成する。検量線の作成は試料測定時に行う。
- 注(5)最適流量は装置によって異なるので、あらかじめ最適条件を求めておく。
- (6)開放送気方式の場合は、還元容器の通気管にコックを付け、塩化すず(Ⅱ)溶液添加後、コックを閉じた状態で約2分間激しく振り混ぜて水銀を気-液平衡に達しさせた後、装置に連結し、ポンプの作動と同時にコックを開く。最適送気速度は先に求めておくが、通常は1~1.5㍑/minである。
- (7)吸光度又はその比例値。開放送気方式の場合はピーク高さを測定する。
- (8)過マンガン酸カリウム溶液(3w/v%)を含む硫酸(1+1)を入れたガス洗浄びんを通して大気中に放出する。
備考 1塩化物イオンを多量に含む試料では、過マンガン酸カリウム処理において塩化物イオンが酸化されて塩素となり、波長253.7nmの光を吸収して正の誤差を生じる。この場合は、塩化ヒドロキシルアンモニウム溶液を過剰に加え、塩素を十分に還元しておく。また、還元容器中に存在する篭素は、窒素などの送入によってあらかじめ追い出しておく。
2 ベンゼン、アセトンなどは253.7nmの光を吸収して正の誤差を生じる。この種の揮発性有機物を含む試験溶液に対しては、過マンガン酸カリウムによる前処理を行った後、次のような操作から適当なものを選び適用する。
- (1)少量のヘキサンと振り混ぜて揮発性有機物を抽出除去する。
- (2)重水素ランプなどによるバックグラウンド補正を行う。
- (3)水銀中空陰極ランプと重水素ランプを用いて指示値の差を求めておき、次に塩化すず(Ⅱ)溶液の添加を省略して同様の測定を行い、両指示値の差から水銀を定量する。
5.1.2 硝酸一流酸一過マンガン酸カリウム分解法
(1)試薬
- (a)ペルオキソニ硫酸カリウム溶液(5w/v%)ペルオキソニ硫酸カリウム(過硫酸カリウム)50gを水に溶かして1㍑とする(注9)。
- (b)その他の試薬は、5.1.1(1)試薬と同じ。
注(9)ぺルオキソニ硫酸アンモニウムを用いてもよい。いずれも溶液中の水銀は0.001mg/㍑以下であること。
(2)器具及び装置
- (a)分解フラスコ 三角フラスコ300ml又はケルダールフラスコ300mlを用いる。
- (b)その他 5.1.1(2)器具及び装置と同じ。
(3)試験溶液の調製
- (a)2.1の湿試料約10gを0.1gのけたまではかり取り、これを分解フラスコに入れ、水を加えて約50mlとする。
- (b)分解フラスコを冷水で冷しながら、硝酸20mlを少しずつ加え静かに混合したのち、硫酸(1+1)20mlを少しずつ加える。
- (c)フラスコ内の反応が止むまで冷水中で放置した後、過マンガン酸カリウム溶液(3w/v%)20mlを加えて振り混ぜ、室温で約15分間放置する。
- (d)過マンガン酸カリウムの色が消えたときは、溶液の赤紫色が15分間持続するまで、過マンガン酸カリウム溶液(3w/v%)を少量ずつ加える。
- (e)ペルオキソニ硫酸カリウム溶液(5w/v%)10mlを加え、約95℃以上の水浴中に分解フラスコの溶液部分を浸して2時間加熱する(注10)。
- (f)以下、5.1.1(3)(d)以後の操作と同様に行う。
注(10)この加熱操作中に過マンガン酸の色が消えた場合は過マンガン酸カリウム溶液(3w/v%)を追加してもよい。
(4)操作
- (a)5.1.1(4)(a)~(d)と同様に操作を行う。
- (b)空試験として分解フラスコに水50mlを入れ(3)(b)~(f)の操作及び(a)の操作について試験溶液と同様に行って指示値を読み、試験溶液について得た指示値を補正する。
検量線 5.1.1(4)検量線と同じ。
5.1.3 硝酸-転化ナトリウム分解法
(1)試薬
- (a)塩化ナトリウム溶液(20w/v%)塩化ナトリウム200gを水を溶かし1㍑とする。
- (b)その他の試薬は、5.1.1(1)試薬と同じ。
(2)器具及び装置
- (a)ケルダールフラスコ300mlを用いる。
- (b)その他は、5.1.2(2)器具及び装置と同じ。
(3)試験溶液の調製
- (a)2.1の湿試料約10gを0.1gのけたまではかり取り、これをケルダールフラスコに入れ、硝酸(1十1)90mlと塩化ナトリウム溶液(20w/v%)20mlを加える。
- (b)約95℃以上の水溶中に、ケルダールフラスコの溶液部分を浸して2時間加熱したのち室温まで冷却する。
- (c)以下5.1.1(3)(e)以後の操作と同様に行う。
(4)操作
- (a)5.1.1(4)(a)~(d)の操作と同様に行う。
- (b)空試験として、ケルダールフラスコに硝酸(1+1)90mlと塩化ナトリウム溶液(20w/v%)20mlを加え、(3)(b)の分解操作及び(a)の操作について試験溶液と同様に行って指示値を読み、試験溶液について得た指示値を補正する。
検量線 水銀標準液(0.1μgHg/ml)1~20mlを還元容器に段階的にとり、硫酸(1+1)10mlと水を加えて約250mlとした後、通気回路を組み立て、5.1.1(4)検量線の操作を行う(注11)。
注(11)溶液中の塩濃度がおおきく異なる場合に、水銀を還元気化したときに、気-液平衡が変化して測定の誤差を生じる場合が有るが、本法によるときは、その誤差は僅かであり無視出来る範囲と判断されている。しかし、無視出来ないことが予想される場合は、検量線作成の溶液についても試料溶液と同一条件になるように、硝酸と塩化ナトリウム溶液を加えて操作を行う。
5.2 アルキル水銀化合物
(1)試薬(注1)
- (a)塩化銅(Ⅰ)粉末
- (b)塩酸
- (c)塩化ナトリウム溶液(20w/v%)
- (d)ベンゼン
- (e)L-システイン-酢酸ナトリウム混合溶液 塩酸L-システイン-水和物1g、酢酸ナトリウム三水和物0.8g及び硫酸ナトリウム(無水)12.8gを水に溶かして100mlとする。
注(1)アルキル水銀化合物を含まないもの。アルキル水銀化合物を含むおそれのある場合は次の操作によりガスクロマトグラムを記録したとき、アルキル水銀化合物の予期保持時間付近にピークを生じないことを確認して用いる。
塩酸 分液漏斗100mlに塩酸10mlと水10ml及びベンゼン5mlを加えて2分間激しく振りまぜ静置する。水層を捨て、ベンゼン層を共栓試験管10mlに移し、硫酸ナトリウム(無水)約1gを加えて振りまぜて脱水する。マイクロシリンジを用いて、このベンゼン5μ㍑を取り(4)の操作に従ってガスクロマトグラフに注入し、ガスクロマトグラムを記録する。
塩化ナトリリム溶液(20w/v%)分液漏斗100mlに塩化ナトリウム溶液(20w/v%)20mlとベンゼン10mlを加えて2分間激しく振りまぜ静置する。水層を捨て、ベンゼン層を共栓試験管10mlに移し、硫酸ナトリウム(無水)約1gを加えて振りまぜて脱水する。以下塩酸と同様に操作してガスクロマトグラムを記録する。
ベンゼン ベンゼン約50mlをロータリーエバポレータ等の濃縮器を用いて約2mlまで濃縮し、マイクロシリンジを用いて、ベンゼン5μ㍑を取り、以下塩酸と同様に操作してガスクロマトグラムを記録する。
L-システイン-酢酸ナトリウム混合溶液分液漏斗20~30mlにL-システイン-酢酸ナトリウム混合溶液8mlと塩酸2ml及びベンゼン5mlを加えて2分間激しく振りまぜ静置する。水層を捨て、ベンゼン層を共栓試験管10mlに移し、硫酸ナトリウム(無水)約1gを加えて振りまぜて脱水する。以下塩酸と同様に操作してガスクロマトグラムを記録する。
- (f)塩化メチル水銀標準液(10mgHg/ml)又は塩化エチル水銀標準液(10mgHg/ml)クロロ(メチル)水銀(Ⅱ)(塩化メチル水銀)0.125g又はクロロ(エチル)水銀(Ⅱ)(塩化エチル水銀)0.132gをそれぞれベンゼンに溶かして全量フラスコ10mlに入れ、ベンゼンを標線まで加える。
- (g)塩化メチル水銀標準液(0.1mgHg/ml)又は塩化エチル水銀標準液(0.1mgHg/ml)塩化メチル水銀標準液(10mgHg/ml)又は塩化エチル水銀標準液(10mgHg/ml)1mlを全量フラスコ100mlにとり、ベンゼンを標線まで加える。
- (h)塩化メチル水銀標準液(0.001mgHg/ml)又は温化エチル水銀標準液(0.001mgHg/ml)塩化メチル水銀標準液(0.1mgHg/ml)又は塩化エチル水銀標準液(0.1mgHg/ml)1mlを全量フラスコ100mlにとり、ベンゼンを標線まで加える。使用時に調製する。
(2)器具及び装置
- (a)分液漏斗 500ml及び20~30mlコックにワセリンなどを使用してはならない。
- (b)共栓試験管 5~10ml
- (c)マイクロシリンジ 1~10μ㍑
- (d)ガスクロマトグラフ
試料導入部温度140~240℃
分離管ガラス製、内径3mm、長さ400~1500mm、温度130~180℃
分離管充てん物 酸洗浄後シラン処理(注2)をしたクロモソルプW(180~250μm)又は同等以上の性能をもつ担体にこはく酸ジエチレングリコール又は同等以上の分離性能をもつ液相を5~25%含浸したもの。
検出器 電子補獲検出器又はこれと同等以上の性能をもつもの、温度140~200℃。
キャリヤーガス 窒素(99.99v/v%以上)、流速30~80ml/min
装置の感度 上記条件で塩化メチル水銀(又は塩化エチル水銀)を水銀(Hg)として0.04μg注入したときのS/N比が3以上であること。
注(2)ジメチルクロロシランのトルエン溶液(1%)中に担体を浸し、水浴上で約1時間保った後、乾燥させる。この処理をした担体が市販されている。また、あらかじめ担体に5~10%の臭化カリウム又は塩化ナトリウムを含浸させた後、液層を被覆したものを用いるとピークが鋭くなる。
(3)試験溶液の調製
- (a)2.1の湿試料約10gを0.1gのけたまではかり取り、共栓付フラスコ300mlに入れ、塩酸(1+1)20mlを加え、ときどき振り混ぜて、硫化水素臭がなくなるまで放置する。
- (b)水30mlと塩化銅粉末100mgを加え約5分間激しく振り混ぜる。
- (c)ベンゼン40mlを加え5分間激しく振り混ぜた後静置する。
- (d)上層のベンゼン層を分離(注3)し、分液漏斗に入れる。
- (e)水層に再びベンゼン40mlを加えて5分間激しく振り混ぜ静置する。
- (d)と同様に操作してベンゼン層を分離する。
- (f)ベンゼン層を合わせ、塩化ナトリウム溶液(20w/v%)20mlを加え、1分間振り混ぜてベンゼン層を洗浄し(注4)、放置後水層を捨てる。
- (g)ベンゼン層にL-システイン-酢酸ナトリウム混合溶液8mlを加え、2分間激しく振り混ぜ、放置後水層を分液漏斗20~30mlに移す。
- (h)水層に塩酸2mlとベンゼン5mlを加えて2分間激しく振り混ぜ、放置後水層を捨て、ベンゼン層を共栓試験管に移す(注5)。これを試験溶液とする。
- 注(3)分離し難いときは、遠心分離器を用いてもよい。遠沈管はふた付のものを用いる。
- (4)多量の無機水銀が存在する場合は電子捕獲検出器を用いたとき、メチル水銀の位置に無機水銀によるピークを生じることがあるので、洗浄を繰り返す。またベンゼン層に塩酸が残留するとL-システインによるアルキル水銀の逆抽出が不完全になるので、洗液が中性になるまで洗浄を繰り返す。
- (5)水分が存在するとガスクロマトグラフに注入したとき異常ピークを生じることがあるので、硫酸ナトリウム(無水)約1gを加えて振り混ぜて脱水する。
(4)操作
- (a)マイクロシリンジを用い、(3)(h)で得た試験溶液の一定量をガスクロマトグラフに注入し、ガスクロマトグラムを記録する。
- (b)塩化メチル水銀又は塩化エチル水銀の保持時間(注6)に相当する位置のピークについて、ピーク面積(又はピーク高さ)を測定する(注7)。
- (c)先に測定に使用した試験溶液1mlを別の共栓試験管にとり、L-システイン-酢酸ナトリウム混合溶液1mlを加えて2分間激しく振り混ぜ、放置する。
- (d)上部のベンゼン層から、先にガスクロマトグラフに注入したベンゼン層と同量をマイクロシリンジを用いてガスクロマトグラフに注入する。
この結果、先に得られたピークが消滅した場合には、先のピークはメチル水銀化合物又はエチル水銀化合物によるものと判定する。 - (e)空試験として塩酸(1+1)20mlをとり、(3)(b)~(h)及び(a)~(d)の操作を行ってピーク面積(又はピーク高さ)を測定し、試験溶液について得たピーク面積(又はピーク高さ)を補正する。
- (f)検量線から水銀の量を求め、試料中のメチル水銀化合物又はエチル水銀化合物を水銀の濃度として算出する。
- (g)3乾燥減量で求めた湿試料の乾燥減量(%)で(f)の濃度を補正し、乾燥試料当りの濃度(mgHg/kg)として結果を表示する。
検量線 分液漏斗20~30mlにL-システイン-酢酸ナトリウム混合溶液8mlをとり、これに塩化メチル水銀標準液(0.001mgHg/ml)又は塩化エチル水銀標準液(0.001mgHg/ml)を検出器の感度に応じて段階的に加え、これに塩酸2mlとベンゼン5mlを加えて2分間激しく振り混ぜ、放置後水層を捨て、ベンゼン層を共栓試験管に移す(注6)。
マイクロシリンジを用い、このベンゼン層の一定量をガスクロマトグラフに注入し、ガスクロマトグラムを記録する。
塩化メチル水銀又は塩化エチル水銀のピークについて、ピーク面積(又はピーク高さ)を測定(注7)して、塩化メチル水銀又は塩化エチル水銀に相当する水銀(Hg)の量とピーク面積(又はピーク高さ)との関係線を作成する。
- 注(6)操作において塩酸を使用するため、メチル水銀化合物又はエチル水銀化合物は、それぞれ塩化メチル水銀又は塩化エチル水銀として挙動する。
- (7)測定時に標準液の一定量を注入して検出器の感度の経時変化を補正する。また、ガスクロマトグラフへの注入量と、得られるピーク面積(又は高さ)との関係が直線となる範囲をあらかじめ求めておき、測定されるピークがこの範囲内となるように注入量を調節する。
6 カドミウム
6.1 原子吸光法
(1)試薬
- (a)カドミウム標準液(0.1mgCd/ml)金属カドミウム(99.9%以上)0.100gを硝酸(1+1)20mlに溶かし、煮沸して窒素酸化物を追い出し、放冷後金量フラスコ1000mlに入れ、水を標線まで加える。又は、日本工業規格(以後、JISと略)K0012(カドミウム標準液)のCd100を用いる。
- (b)カドミウム標準液(0.01mgCd/ml)カドミウム標準液(0.1mgCd/ml)50mlを全量フラスコ500mlにとり、硝酸(1+1)10mlを加えた後、水を標線まで加える。又は、JIS K0012のCd10を用いる。
(2)試験溶液の調製
- (a)2.3の乾燥試料の適量(注1)(2~5g程度)をビーカー200mlに10mgのけたまではかり取る。
- (b)硝酸10mlと塩酸20mlを加え、軽く振って試料と酸を混和させた後、熱板上で加熱する。加熱中は、時計皿でふたをする(注2)。
- (c)液量が約半分になったら一旦ビーカーを熱板からおろし、硝酸20mlを加え、再び同様に加熱を続け、液量が20mlになったら放冷する。
- (d)ビーカーの壁を少量の水で洗い、水50mlを加えて静かに加熱した後、不溶解物が沈降するのをまって、ろ紙5種Bでろ過し、ろ液を別のビーカー200mlに入れる。
- (e)先のビーカー中の不溶解物を少量の塩酸(1+10)で洗浄し、洗液を(d)のろ紙上に移し入れる。この操作を2~3回繰り返す。
- (f)ろ液と洗液(注3)を入れたビーカーを熱坂上で液量が2~3mlになるまで加熱し、放冷する。
- (g)ビーカーの壁の付着物を少量の水で洗い落とし、塩酸(1+10)10mlを加えて加熱し、析出物を溶解する(注4)。
- (h)放冷後、全量フラスコ100mlに移し入れ、更に少量の水でビーカーを洗って同様に移し入れた後、水を標線まで加え、これを試験溶液とする。
- 注(1)2.1の湿試料又は2.2の風乾試料を用いてもよい。
- (2)分解にともなう反応が止んだら時計皿は少しずらすか、ガラス棒を用いるなど適当な方法でうかしておく。
- (3)ろ液と洗液を合わせた液が黒褐色~褐色を呈するときは、これに硝酸10mlと過塩素酸5mlを加え、熱坂上で課塩素酸の白煙が発生するまで加熱する。ただし、過塩素酸の白煙が発生したときに、液がまだ黒褐色~褐色の場合は直ちにビーカーを熱板からおろし、放冷後、硝酸10mlを加えて再び加熱する。この操作を、液が淡黄色~無色を呈するまで繰り返す。引き続き加熱して大部分の過塩素酸を除去し、放冷後、(g)以下の操作を行う。
- (4)この時、有機物が残存し、内容物があめ状になったり析出物が生じたりして、これらが塩酸(1+10)10mlを加えて加熱しても溶解しない場合がある。このときは、放冷後、硝酸5mlを加え、次いで過酸化水素水(30%)3~4mlを徐々に加え、激しい泡立ちが止んだら熱坂上で液量が2~3mlになるまで加熱し、以後(g)以下の操作を行う。
(3)操作
- (a)試験溶液の適量(Cdとして0.005~0.2mgを含む)を全量フラスコ100mlにとり、塩酸(1+10)10mlを加えた後、水を標線まで加える。
- (b)バックグラウンド補正装置付原子吸光分析装置で、波長228.8nmにおける指示値(吸光度又はその比例値)を読む。
- (c)空試験として、(2)(b)~(h)の操作を行ったものについて試験溶液の場合と同様に(a)~(b)の操作を行って指示値を読み、(b)の指示値を補正する。
- (d)検量線からカドミウム量を求める。
- (e)乾操試料当りのカドミウム濃度(mgCd/kg)として結果を表示する。
検量線 カドミウム標準液(0.01mgCd/ml)0.5~20mlを全量フラスコ100mlに段階的にとり、塩酸(1+10)10mlを加え、水を標線まで加えた後、波長228.8nmにおける指示値を読み、カドミウム量と指示値との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
6.2 溶媒抽出一原子吸光法
(1)試薬
- (a)ジエチルジチオカルバミン酸ナトリウム溶液(5w/v%) ジエチルジチオカルバミン酸ナトリウム三水和物6.5gを水に溶かして100mlとし、着色びんに保存する。調製後、1週間以上経過したものを使用してはならない。
- (b)くえん酸水素アンモニウム溶液(20w/v%)くえん酸水素アンモニウム(くえん酸ニアンモニウム)20gを水に溶かし100mlとする。くえん酸水素アンモニウム中にカドミウム等の不純物が含まれる恐れがあるときは、次の操作によって精製する。
くえん酸水素アンモニウム20gを水80mlに溶かし、アンモニア水(1+1)を加えてpH約9とした後、水を加えて100mlとする。これを分液漏斗に入れ、ジエチルジチオカルバミン酸ナトリウム溶液(1w/v%)2ml及び酢酸プチル10mlを加え、激しく振り混ぜて放置する。水層を乾いたろ紙でろ過し、酢酸プチルの微泡をのぞいたろ液を用いる。 - (c)カドミウム標準液(0.01mgCd/ml)6.1(1)(b)と同し。
(2)試験溶液の調製
(a)6.1(2)(a)~(h)の操作を行い、試験溶液を調製する。
(3)操作
- (a)試験溶液の適量(Cdとして0.001~0.05mgを含む)を分液漏斗200mlにとり、くえん酸水素アンモニウム溶液(20w/v%)10mlを加え、アンモニア水(1+1)を用いてpH9~9.5に調節する(注5)。
- (b)ジエチルジチオカルバミン酸ナトリウム溶液(5w/v%)10mlを加え、水で全容を約150mlとして軽く振り混ぜた後、酢酸プチル10mlを加えて2~3分間激しく振り混ぜる。
- (c)静置して水層と酢酸プチル層とを十分分離した後、水層は別の分液漏斗200mlに入れ、酢酸プチル層はビーカー50mlに入れる(注6)。
- (d)水層を入れた分液漏斗に酢酸プチル10mlを加えて2~3分間激しく振り混ぜる。
- (e)静置後、水層は捨て、酢酸プチル層は先のビーカー50mlに入れる。
- (f)分液漏斗は少量の酢酸プチルで洗い、これを先のビーカー50mlに入れる。
- (g)酢酸プチル層を入れたビーカーを熱坂上で静かに加熱して、酢酸プチルを揮散させる(注7)。
- (h)放冷後、硝酸4mlと過塩素酸2mlを加え、熱坂上で静かに加熱して有機物(ジエチルジチオカルバミン酸錯体)を酸化分解し、次いで蒸発乾固する。
- (i)塩酸(1+10)5mlを加え、加熱して析出物を溶解して全量フラスコ25ml(注8)に移し入れ、更に少量の水でビーカーを洗って同様に移し入れた後、水を標線まで加える。
- (j)原子吸光分析装置で、波長228.8nmにおける指示値を読む。
- (k)空試験として、6.1(2)(b)~(h)の操作を行ったものについて、試験溶液の場合と同様に(a)~(j)の操作を行って指示値を読み、(j)で得た指示値を補正する。
(1)検量線からカドミウム量を求める。
- (m)乾燥試料当りのカドミウム濃度(mgCd/kg)として結果を表示する。
検量線 カドミウム標準液(0.01mgCd/ml)0.5~20mlを全量フラスコ100mlに段階的にとり、塩酸(1+10)10mlを加え、水を標線まで加えた後、波長228.8nmにおける吸光度又はその比例値を測定し、カドミウム量と指示値との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
- 注(5)フェノールフタレイン指示薬(フェノールフタレイン1.0gをエタノール90mlに溶解、水で100mlとしたもの)を用いるとよい。指示薬2~3滴を加えた後、アンモニア水(1+1)を液が紅色を呈するまで加える。変色点が見難い場合はpH計又はpH試験紙を用いる。
- (6)酢酸プチル層に水分が混入しないように操作する。水分が混入すると(g)の加熱時に突沸することがある。(e)及び(f)の場合もこれと同じように操作する。
- (7)酢酸プチルは完全に揮散させる。酢酸プチルが残留すると、(h)の有機物の酸化分解が不十分になる。
- (8)全量フラスコ10mlを用いてもよい。ただし、その場合は(a)の試験溶液の適量としてCd0.0005~0.02mgを含む量を分液漏斗200mlにとる。
7 鉛
7.1 原子吸光法
(1)試薬
(a)鉛標準液(0.1mgPb/ml)金属鉛(99.9%以上)0.100gを硝酸(1+3)40mlに溶かし、煮沸して窒素酸化物を追い出し、冷却後全量フラスコ1000mlに入れ、水を標線まで加える。又は、JIS K0015(鉛標準液)のPb100を用いる。
(2)試験溶液の調製
(a)6.1(2)(a)~(h)の操作を行い、試験溶液を調製する。
(3)操作
- (a)試験溶液の適量(Pbとして0.1~2mgを含む)を全量フラスコ100mlにとり、塩酸(1+10)10mlを加えた後、水を標線まで加える。
- (b)バックグラウンド補正装置付原子吸光分析装置で、波長283.3nmにおける指示値(吸光度又はその比例値)を読む。
- (c)空試験として、6.1(b)~(h)までの操作を行ったものについて、試験溶液の場合と同様に(a)~(b)の操作を行って指示値を読み、(b)の指示値を補正する。
- (d)検量線から鉛量を求める。
- (e)乾燥試料当りの鉛濃度(mgPb/kg)として結果を表示する。
検量線 鉛標準液(0.1mgPb/ml)1~20mlを全量フラスコ100mlに段階的にとり、塩酸(1+10)10mlを加え、水を標線まで加えた後、波長283.3nmにおける指示値を読み、鉛量と指示値との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
7.2 溶媒抽出一原子吸光法
(1)試薬
- (a)ジエチルジチオカルバミン酸ナトリウム溶液(5w/v%)6.2(1)(a)と同じ。
- (b)くえん酸水素アンモニウム溶液(20w/v%)6.2(1)(b)と同じ。
- (c)鉛標準液(0.1mgPb/ml)7.1(1)(a)と同し。
(2)試験溶液の調製
(a)6.1(2)(a)~(h)の操作を行い、試験溶液を調製する。
(3)操作
- (a)試験溶液の適量(Pbとして0.025~0.5mgを含む)を分液漏斗200mlにとり、6.2(3)(a)~(i)の操作を行う(注1)。
- (b)原子吸光分析装置で、波長283.3nmにおける指示値を読む。
- (c)空試験として、6.1(2)(b)~(h)の操作を行ったものについて、試験溶液の場合と同様に(a)~(b)の操作を行って指示値を読み、(b)の指示値を補正する。
- (d)検量線から鉛量を求める。
- (e)乾燥試料当りの鉛濃度(mgPb/kg)として結果を表示する。
検量線 鉛標準液(0.1mgPb/ml)-1~20加を全量フラスコ100mlに段階的にとり、塩酸(1+10)10mlを加え、水を標線まで加えた後、波長283.3nmにおける指示値を読み、鉛量と指示値との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
注(1)最終段階で10mlとする場合は、(a)の試験溶液の適量としてPb0.01~0.2mgを含む量を分液漏斗200mlにとる。
8 銅
8.1 原子吸光法
(1)試薬
- (a)鋼標準液(0.1mgCu/ml) 金属銅(99.9%)0.100gを硝酸(1+1)20mlに溶かし、煮沸して窒素酸化物を追い出し、放冷後全量フラスコ1000mlに入れ、水を標線まで加える。又は、JIS K0010(銅標準液)のCu100Oを用いる。
- (b)銅標準液(0.01mgCu/ml)銅標準液(0.1mgCu/ml)50mlを全量フラスコ500mlにとり、硝酸(1+1)10mlを加え、水を標線まで加える。又は、JIS K001.0(銅標準液)のCu10を用いる。
(2)試験溶液の調製
(a)6.1(2)(a)~(h)の操作を行い、試験溶液を調製する。
(3)操作
- (a)試験溶液の適量(Cuとして0.02~0.4mgを含む)を全量フラスコ100mlにとり、塩酸(1+10)10mlを加えた後、水を標線まで加える。
- (b)原子吸光分析装置で、波長324.7nmにおける指示値(吸光度又はその比例値)を読む。
- (c)空試験として、6.1(2)(b)~(h)の操作を行ったものについて、試験溶液の場合と同様に(a)~(b)の操作を行って指示値を読み、(b)で得た指示値を補正する。
- (d)検量線から銅量を求める。
- (e)乾燥試料当りの銅濃度(mgCu/kg)として結果を表示する。
検量線 銅標準液(0.01mgCu/ml)2~40mlを全量フラスコ100mlに段階的にとり、塩酸(1+10)10mlを加え、水を標線まで加えた後、波長324.7nmにおける指示値を読み、銅量と指示値との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
9 亜 鉛
9.1 原子吸光法
(1)試薬
- (a)亜鉛標準液(0.1mgZn/ml) 金属亜鉛(99.9%以上)0.100gを硝酸(1+1)20mlを溶かし、煮沸して窒素酸化物を追い出し、放冷後全量フラスコ1000mlに入れ、水を標線まで加える。又は、JISK0011(亜鉛標準液)のZn100を用いる。
- (b)亜鉛標準液(0.01mgZn/ml) 亜鉛標準液(0.1mgZn/mlの50mlを全量フラスコ500mlにとり、硝酸(1+1)10mlを加え、水を標線まで加える。又は、JIS K0011(亜鉛標準液)のZn10を用いる。
(2)試験溶液の調製
(a)6.1(2)(a)~(h)の操作を行い、試験溶液を調製する。
(3)操作
- (a)試験溶液の適量(Znとして0.005~0.2mgを含む)を全量フラスコ100mlにとり、塩酸(1+10)10mlを加えた後、水を標線まで加える。
- (b)原子吸光分析装置で、波長213.9nmにおける指示値(吸光度又はその比例値)を読む。
- (c)空試験として、6.1(2)(b)~(h)の操作を行って指示値を読み、(b)で得た指示値を補正する。
- (d)検量線から亜鉛量を求める。
- (e)乾燥試料当りの亜鉛濃度(mgZn/kg)として結果を表示する。
検量線 亜鉛標準液(0.01mgZn/ml)0.5~20mlを全量フラスコ100mlに段階的にとり、塩酸(1+10)10mlを加え、水を標線まで加えた後、波長213.9nmにおける指示値を読み、亜鉛量と指示値との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
10 鉄
10.1原子吸光法
(1)試薬
- (a)鉄標準液(1mgFe/ml) 金属鉄(99.9%以上)1.00gを塩酸(1+1)30mlに加熱して溶かし、放冷後全量フラスコ1000mlに入れ、水を標線まで加える。又は、硫酸鉄(Ⅱ)アンモニウム六水和物7.02gを塩酸(1+1)20mlと適量の水に溶かし、全量フラスコ1000mlに入れ、水を標線まで加える。又は、JIS K0016(鉄標準液)のFe1000を用いる。
- (b)鉄標準液(0.01mgFe/ml) 鉄標準液(1mgFe/ml)5mlを全量フラスコ500mlにとり、塩酸(1+1)10mlを加え、水を標線まで加える。又は、JIS K0016(鉄標準液)のFe10を用いる。
(2)試験溶液の調製
(a)6.1(2)(a)~(h)の操作を行い、試験溶液を調製する。
(3)操作
- (a)試験溶液の適量(Feとして0.03~0.6mgを含む)を全量フラスコ100mlにとり、塩酸(1+10)10mlを加えた後、水を標線まで加える。
- (b)原子吸光分析装置で、波長248.3nmにおける指示値(吸光度又はその比例値)を読む。
- (c)空試験として、6.1(2)(b)~(h)の操作を行ったものについて、試験溶液の場合と同様に(a)~(b)の操作を行って指示値を読み、
- (b)の指示値を補正する。
- (d)検量線から鉄量を求める。
- (e)乾燥試料当りの鉄濃度(mgFe/kg)として結果を表示する。
検量線 鉄標準液(0.01mgFe/ml)3~60mlを全量フラスコ100mlに段階的にとり、塩酸(1+10)10mlを加え、水を標線まで加えた後、波長248.3nmにおける指示値を読み、鉄量と指示値との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
11 マンガン
11.1原子吸光法
(1)試薬
- (a)マンガン標準液(0.1mgMn/ml)金属マンガン(99.9%以上)0.100gを硫酸(1+3)20mlに加熱して溶かし、放冷後全量フラスコ1000mlに入れ、水を標線まで加える。又は、過マンガン酸カリウム0.290gをとり、水150mlに硫酸(1+1)10mlを加えた溶液に溶かし、亜硫酸水素ナトリウム溶液(10w/v%)を滴加し、かき混ぜて脱色させた後、煮沸して過剰の亜硫酸を追い出し、放冷後全量フラスコ1000mlに入れ、水を標線まで加える。又は、JIS K0027(マンガン標準液)のMn100を用いる。
- (b)マンガン標準液(0.01mgMn/ml) マンガン標準液(0.1mgMn/ml)50mlを全量フラスコ500mlにとり、硝酸(1+1)10mlを加え、水を標線まで加える。又は、JIS K0027(マンガン標準液)のMn10を用いる。
(2)試験溶液の調製
(a)6.1(2)(a)~(h)の操作を行い、試験溶液を調製する。
(3)操作
- (a)試験溶液の適量(Mnとして0.01~0.4mgを含む)を全量フラスコ100mlにとり、塩酸(1+10)10mlを加えた後、水を標線まで加える。
- (b)原子吸光分析装置で、波長279.5nmにおける指示値(吸光度又はその比例値)を読む。
- (c)空試験として、6、1(2)(b)~(h)の操作を行ったものについて、試験溶液の場合と同様に(a)~(b)の操作を行って指示値を読み、(b)の指示値を補正する。
- (d)検量線からマンガン量を求める。
- (e)乾燥試料当りのマンガン濃度(mgMn/kg)として結果を表示する。
棟量線 マンガン標準液(0.01mgMn/ml)1~40mlを全量フラスコ100mlに段階的にとり、塩酸(1+10)10mlを加え、水を標線まで加えた後、波長279.5nmにおける指示値を読み、マンガン量と指示値との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
12 クロム(総クロム、酸溶出クロム、6価クロム)
12.1総クロム
12.1.1 炭酸ナトリウム融解-吸光光度法
(1)試薬
- (a)過マンガン酸カリウム溶液(3w/v%)過マンガン酸カリウム3gを水に溶かし100mlとする。
- (b)尿素溶液(20w/v%)尿素20gを水に溶かし100mlとする。
- (c)亜硝酸ナトリウム溶液(2w/v%)亜硝酸ナトリウム2gを水に溶かし100mlとする。使用時に調製する。
- (d)ジフェニルカルバジド溶液(1w/v%)1.5-ジフェニルカルボノヒドラジド(ジフェニルカルバジド)0.5gをアセトン25mlに溶かし、水を加えて50mlとする。冷暗所に保存する。保存期間は、約1週間である。
- (e)クロム標準液(1mgCr/ml)二クロム酸カリウムを100~110℃で3~4時間乾燥し、デシケーター中で放冷した後、その0.283gを水に溶かして全量フラスコ100mlに入れ、水を標線まで加える。
又は、JIS K0024(クロム標準液)のCr1000を用いる。 - (f)クロム標準液(0.01mgCr/ml)クロム標準液(1mgCr/ml)の1.0mlを全量フラスコ100mlにとり、水を標線まで加える。又は、JISK0024(クロム標準液)のCr10を用いる。
(2)試験溶液の調製
- (a)2.3の乾燥試料をめのう製乳ばちを用いて細かくすりつぶし、その1.0gを10mgのけたまで磁器製のるつぼにはかり取り、電気炉で徐々に温度を上げ550℃で2時間灰化(注1)する。
- (b)るつぼの内容物を白金るつぼ(内容量20~30ml)に移し入れる。
- (c)これに硫酸(1+2)数滴とふっ化水素酸(2)20mlを加えドラフト内において熱板上で硫酸白煙が発生し始めるまで加熱する。
- (d)放冷した後、ふっ化水素酸5mlを加え、硫酸白煙の発生が殆んどなくなるまで加熱する。
- (e)引き続き白金るつぼを直火で徐々に温度を上げ、硫酸白煙が発生しなくなるまで加熱し放冷する。
- (f)白金るつぼに炭酸ナトリウム5g及び硝酸ナトリウム0.3gを加えよく混合しふたをした後、直火で徐々に温度を上げ、約900℃で時々るつぼをゆり動かし、内容物をよく混ぜ合わせ、約20分間加熱融解する。
- (g)放冷後、白金るつぼに温水を加え融解物(注3)をビーカー200mlに移し入れる。
- (h)ビーカーを水浴上で加温してクロム酸塩を浸出する。これをろ紙5種Bを用いてろ過(注4)し、ろ紙上の沈激物を温水で洗浄する(注5)。
- (i)ろ液と洗液を合わせ、硫酸(1+2)を加えて中和する。これを蒸発し(注6)、冷却後、全量フラスコ100mlに移し入れ、水を標線まで加え、これを試験溶液とする。
- (j)別に分析試料を入れない白金るつぼ等を用いて(c)~(i)の操作(注7)を行い空試験液とする。
- 注(1)分析試料の灰化操作は、試料中の有機物の灰化を目的としたもので、本操作により、アルカリ融解に際し、有機物分解による激しい反応によって生ずる融解物の損失を防ぐと共に白金るつぼの損傷を避けることができる。
- (2)灰化の終った試料についてのふっ化水素酸処理及び融解操作は、けい酸の揮散除去により後に行う融解反応の促進をはかるものである。硫酸白煙を発生させる際の加熱は、突沸させないよう注意し、熱板の濃度に配慮して行う。
- (3)アルカリ融解物を温水を用いて白金るつぼから取り出すのが困難な場合は、白金るつぼとふたを温水約50mlを加えたビーカー200mlに入れ、(h)の操作を行う。ろ過に先立ってるつぼとふたは水洗して取り出しておく。洗液はビーカーに加える。
- (4)鉄を除くろ過操作に長時間をかけると、クロムが3価に還元され、水酸化鉄の沈毅へ吸着されるため負の誤差の原因となる。これを防ぐためアルカリ融解物の温浸液が温かい状態(70~80℃)でろ過するのがよい。
- (5)不溶解物中にクロム分が残存する恐れのあるときには、不溶解物はろ紙ごと乾燥したのち再灰化処理を行い、この灰分について融解操作を換り返す。
- (6)液量が100mlを超える場合には濃縮操作が必要であるが、ろ液と洗液を合わせても80ml以下であれば、この操作を行う必要はない。
- (7)このとき(f)の加熱融解は、るつぼの内容物が融解状態となったところまででよい。
(3)操作
- (a)試験溶液の適量(Crとして0.015~0.25mgを含む)をビーカー100mlにとり、これに硫酸全量が2~3ml(注8)になるように硫酸(1+2)6~9mlを加え、数分間煮沸したのち冷却し、全量フラスコ50mlに洗い移し、水を標線まで加える。この溶液から20mlをビーカー100mlに分取する。
- (b)ビーカーに過マンガン酸カリウム溶液(3w/v%)を液の色が赤紫色になるまで滴加し、更に2~3滴加え、静かに数分間煮沸してクロムを完全に酸化する(加熱中に液の赤紫色が消えそうになったら、過マンガン酸カリウムを滴加し、常時液の色を赤紫色に保っておく)。
- (c)室温まで冷却した後、尿素溶液(20w/v%)10mlを加え激しくかき混ぜながら亜硝酸ナトリウム溶液(2w/v%)を液の赤紫色が消えるまで1滴ずつ加える(注9)。
- (d)少量の水で全量フラスコ50mlに移し入れ、過剰の亜硝酸と尿素の反応による泡が消えるまで振り混ぜる。
- (e)液温を20℃以下に冷却した後、ジフェニルカルバジド(1w/v%)3mlを加え直ちに振り混ぜ、水を標線まで加え、更に振り混ぜて発色させる。
- (f)室温で10分間放置後(注10)、この溶液の一部を吸収セル(10mm)に移し、波長540nm付近の吸光度を測定する。
- (g)(2)(j)の空試験液を用いて試験溶液と同様に(a)~(f)の操作を行って吸光度を求め、(f)の吸光度を補正する。
- (h)検量線からクロム量を求める。
- (i)乾燥試料当りのクロム濃度(mgCr/kg)として結果を表示する。
検量線 クロム標準液(0.01mgCr/ml)を1.5~25mlをビーカー100mlに段階的にとり、水で約25mlとし、硫酸(1+2)2mlを加え、数分間煮沸したのち冷却し、全量フラスコ50mlに洗い移し、水を標線まで加える。この溶液から20mlをビーカー100mlにとり、以下、(b)~(f)の操作を行って、クロム量と吸光度との関係線を作成する。
- 注(8)ジフェニルカルバジドによるクロム(Ⅵ)の発色時の硫酸の添加が大きすぎないように注意する。
- (9)亜硝酸ナトリウムによる過マンガン酸カリウムの分解に際して、クロム(Ⅵ)が還元され負の誤差の原因となることがある。必ずよくかき混ぜながら亜硝酸ナトリウム溶液(2w/v%)を滴加する。
- (10)ジフェニルカルバジドによるクロムの定量に対し影響する防害元素としてバナジウム等があるが、呈色後10分間の放置によりその影響はなくなる。
12.1.2 炭酸ナトリウム融解-溶媒抽出-原子吸光法
(1)試薬
- (a)過マンガン酸カリウム溶液(3w/v%)12.1.1(1)(a)と同じ。
- (b)トリオクチルアミンー酢酸プチル溶液(3v/v%)トリオクチルアミン3mlを酢酸プチルに溶かし100mlとする。
- (c)クロム標準液(1mgCr/ml)12.1.1(1)(e)と同じ。
- (d)クロム標準液(0.01mgCr/ml)12.1.1(1)(f)と同じ。
(2)試験溶液の調製
(a)12.1.1(2)(a)~(j)の操作を行い、試験溶液及び空試験液を調製する。
(3)操作
- (a)試験溶液の適量(Crとして、0.01~0.1mgを含む)をビーカー100mlにとり硫酸(1+2)2mlを加え数分間煮沸した後、過マンガン酸カリウム溶液(3w/v%)を液の色が赤紫色になるまで滴加し、更に2~3滴を加えた後数分間煮沸してクロムを完全に酸化する(加熱中に液の赤紫色が消えそうになったら、過マンガン酸カリウム溶液を滴加し、常時、液の色を赤紫色に保っておく)。
- (b)室温まで冷却した後、分液漏斗200mlに移し、水を加えて100mlとする。
- (c)分液漏斗にトリオクチルアミンー酢酸プチル溶液(3v/v%)10mlを加えて10分間振り混ぜる。
- (d)酢酸プチル層をとり(乾燥ろ紙でろ過するなどして水分を取り除いておく)、この液を原子吸光分析装置を用いて波長357.9nmにおける指示値(吸光度又はその比例値)を読む。
- (e)(2)(a)の空試験液を用い試験溶液と同時に(a)~(d)の操作を行って指示値を読み、(d)の指示値を補正する。
- (f)検量線からクロム量を求める。
- (g)乾燥試料当りのクロム濃度(mgCr/kg)として結果を表示する。
検量線 クロム標準波(0.01mgCr/ml)1~10mlを段階的にとり、(a)~(d)の操作を行って、クロム量と指示値との関係線を作成する。
12.1.3 過酸化ナトリウム融解-吸光光度法
(1)試薬
- (a)過マンガン酸カリウム溶液(3w/v%)12.1.1(1)(a)と同し。
- (b)尿素溶液(20w/v%)12.1.1(1)(b)と同じ。
- (c)亜硝酸ナトリウム溶液(2W/V%)12.1.1(1)(c)と同じ。
- (d)ジフェニルカルバジド溶液(1W/V%)12.1.1(1)(d)と同じ。
- (e)クロム標準液(1mgCr/ml)12.1.1(1)(e)と同じ。
- (f)クロム標準液(0.01mgCr/ml)12.1.1(1)(f)と同じ。
(2)試験溶液の調製
- (a)2.3の乾燥試料をめのう製乳ばちを用いて細かくすりつぶし、その0.5gをニッケルるつぼ(注11)に入れ、電気炉で徐々に温度を上げ550℃で2時間灰化する。
- (b)放冷後、過酸化ナトリウム約5gを入れて混合し、さらに少量の過酸化ナトリウムで表面をおおい、バーナー直火で初めは徐々に加熱し、内容物が融解状となってから温度を高め、約3分間赤熱状(あまり高温にしない)として融解後放冷する。
- (c)るつぼを50mlの水を加えたビーカー300mlに入れる。温水50mlを注意しながら少しずつ加え、加熱してるつぼの内容物を浸出する。
- (d)るつぼを水で洗って取り出す。浸出液をかき混ぜながら過酸化ナトリウムを少量ずつ加えて加熱煮沸して、クロムを完全にクロム(Ⅵ)に酸化するとともに過剰の過酸化ナトリウムを分解する。
- (e)室温まで放冷後、全量フラスコ250mlに沈殿物ごと移し入れ、水を標線まで加えてよく振り混ぜたのち静置する。
- (f)上澄液をろ紙5種Bでろ過し、初めのろ液10mlは捨て、次のろ液を試験溶液とする。
- (g)別に試料を入れないニッケルるつぼ等について(b)~(f)の操作を行い、空試験液とする。
(3)操作
- (a)12.1.1(3)(a)~(h)の操作を行う。
- (b)乾燥試料当りのクロム濃度(mgCr/kg)として結果を表示する。
検量線12.1.1(3)の検量線と同じ。
注(11)ニッケルのほか、鉄、アルミナ又はジルコニア製のるつぼを用いることができる。
12.1.4 過酸化ナトリウム融解-溶媒抽出-原子吸光法
(1)試薬
- (a)過マンガン酸カリウム溶液(3w/v%)12.1.1(1)(a)と同じ。
- (b)トリオクチルアミンー酢酸プチル溶液(3v/v%)12.1.2(1)(b)と同じ。
- (c)クロム標準液(1mgCr/ml)12.1.1(1)(e)と同じ。
- (d)クロム標準液(0.01mgCr/ml)12.1.1(1)(f)と同じ。
(2)試験溶液の調製
(a)12.1.3(2)(a)~(g)の操作を行い、試験溶液及び空試験液を調製する。
(3)操作
(a)12.1.3(2)(a)~(g)の操作を行う。
検量線12.1.2(3)の検量線と同じ。
12.2 酸溶出クロム
12.2.1 原子吸光法
(1)試薬
- (a)クロム標準液(1mgCr/ml)12,1.1(1)(e)と同じ。
- (b)クロム標準液(0.01mgCr/ml)12.1.1(1)(f)と同じ。
(2)試験溶液の調製
- (a)6.1(2)(a)~(h)の操作を行い、試験溶液を調製する。
- (b)空試験として、6.1(2)(b)~(h)の操作を行って空試験液を調製する。
(3)操作
- (a)試験溶液の適量(Crとして、0.01~0.1mgを含む)を全量フラスコ100mlにとり、塩酸(1+10)10mlを加えたのち、水を標線まで加える。
- (b)(a)の液について空気-アセチレンフレーム又は酸化二窒素-アセチレンフレーム(12)を用い、原子吸光分析装置で波長357.9nmの指示値(吸光度又はその比例値)を読む。
- (c)(2×b)の空試験について(a)~(b)の操作を行って(b)の指示値を補正する。
- (d)検量線からのクロム量を求める。
- (e)乾燥試料当りのクロム濃度(mgCr/kg)として結果を表示する。
検量線 クロム標準液(0.01mgCr/ml)1~10mlを段階的にとり、(a)~(b)の操作を行って、クロム量と指示値の関係線を作成する。
注(12)空気-アセチレンフレームでは、多燃料フレームにすると感度は高くなるが、鉄、ニッケルなど共存物による妨害も大きくなる。この場合、硫酸ナトリウム、二硫酸カリウム異はニふっ化水素アンモニウムを%程度度共存させる。酸化二窒素-アセチレンフレームでは妨害の大部分は消滅する。
12.3 六価クロム
12.3.1 吸光光度法
(1)試薬
- (a)ジフェニルカルバジド溶液(1w/v%)12.1.1(1)(d)と同じ。
- (a)クロム標準液(1mgCr/ml)12.1.1(1)(e)と同じ。
- (b)クロム標準液(0.01mgCr/ml)12.1.1(1)(f)と同じ。
(2)試験溶液の調製
- (a)混合液に含まれる乾燥固型分と混合液の重量体積比が3/100になり、かつ、混合液量が500ml以上になるように、2.1の湿試料をとり、水を加えて混合液を調製し、室温において4時間連続して振り混ぜる。
- (b)30分放置した後、ろ紙5種Bを用いてろ過しこれを試験溶液とする。
(3)操作
- (a)試験溶液の適量(Cr(Ⅵ)として0.015~0.2mgを含む)を全量フラスコ50mlにとり、硫酸(1十1)0.5~0.6mlを加えて振り混ぜた後、液温を20℃以下に冷却する。
- (b)これにジフェニルカルバジド溶液(1w/v%)0.5mlを加え、直ちに振り混ぜ水を加え、更に振り混ぜて発色させる。
- (c)室温で約10分放置後、この一部を吸収セルに移し、(d)の液を対照液として波長540nm付近の吸光度を測定する。
- (d)別にビーカー100mlに、(a)で分取したのと同量の試験溶液をとり、硫酸(1+2)0.5~0.6mlを加え、エタノール(95)数滴を加え煮沸してクロム酸を還元し放冷する。冷却後、全量フラスコ50mlに入れ、これにジフェニルカルバジド溶液(1w/v%)0.5mlを加え、水を標線まで加えて振り混ぜ10分間放置する。
- (e)検量線から試験溶液中のクロム量を求め、これから乾燥試料中の六価クロム濃度(mgCr(Ⅵ)/kg)を算出する。
検量線12.1.1(3)の検量線と同じ。
12.3.2 溶媒抽出-原子吸光法
(1)試薬
- (a)トリオクチルアミン-酢酸プチル溶液(3v/v%)12.1.2(1)(b)と同じ。
- (b)クロム標準液(0.01mgCr/ml)12.1.1(1)(f)と同じ。
(2)試験溶液の調製
(a)12.3.1(2)(a)~(b)の操作を行い、試験溶液を調製する。
(3)操作
- (a)試験溶液の適量(Crとして0.01~0.10mgを含む)を全ビーカー200mlにとり、水を加えて100mlとする。
- (b)pH計を用い硫酸(3+50)を加えて中和し、更に硫酸(3+50)20mlを加え、分液漏斗200mlに移し、水を加えて200mlとする。
- (c)分液漏斗にトリオクチルアミンー酢酸プチル溶液(3v/v%)20mlを加えて10分間振り混ぜる。
- (d)酢酸ブチル層をとり(乾燥ろ紙でろ過するなどして水分を取り除いておく)、この液を原子吸光分析装置を用い波長357.9nmでの指示値(吸光度又はその比例値)を読む。
- (e)空試験として、分液漏斗200mlに硫酸(3+50)20mlを入れ、水を加えて200mlとし、(c)~(d)の操作を行って指示値を読み、(d)の指示値を補正する。
- (f)検量線から試験溶液中のクロム量を求め、これから乾燥試料中の六価クロム濃度(mgCr(Ⅵ)/kg)を算出する。
検量線12.1.2(3)の検量線と同じ。
13 ひ素
13.1 ジエチルジチオカルバミン酸銀吸光光度法
(1)試薬
- (a)よう化カリウム溶液(20w/v%)よう化カリウム20gを水に溶かして100mlとする。
- (b)塩化すず(Ⅱ)溶液 塩化すず(Ⅱ)二水和物40gを塩酸に溶かし、塩酸で100mlとする。小粒のすず2~3個を加え褐色びんに入れて保存する。使用時に水で10倍に薄める。
- (c)酢酸鉛溶液(10w/v%) 酢酸鉛三水和物12gを酢酸1~2滴と水に溶かして100mlとする。
- (d)亜鉛 JIS K8012〔亜鉛(試薬)〕のひ素分析用(砂状)(1000~1410μm)
- (e)ジエチルジチオカルバミン酸銀溶液 N,N一ジエチルジチオカルバミン酸銀0.25gとブルシンニ水和物0.1gをクロロホルムに溶かして100mlとし、暗所に保存する(注1)。
- (f)ひ素標準液(0.1mgAs/ml) 三酸化ひ素0.132gを水酸化ナトリウム溶液(4w/v%)2mlに溶かした後、水を加えて500mlとし、次に硫酸(1+10)を加えて徴酸性とする。これを全量フラスコ1000mlに移し、水を標線まで加える。又は、JIS K0026(ひ素標準液)のAs100を用いる。
- (g)ひ素標準液(0.001mgAs/ml) ひ素標準液(0.1mgAs/ml)10mlを全量フラスコ1000mlにとり、水を標線まで加える。使用時に調製する。又は、JIS K0026(ひ素標準液)のAs1を用いる。
注(1)ジエチルジチオカルバミン酸銀のピリジン溶液(0.5w/v%)を用いてもよい。
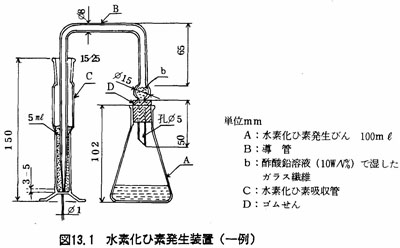
(2)装置
(a)水素化ひ素発生装置 図13.1に示す。
(3)試験溶液の調製
- (a)2.1の湿試料の適量(注2)(乾燥物質量として約2g程度)をビーカー200mlに10mgのけたまではかり取る。
- (b)硝酸15ml、硫酸(1+1)15mlを加え、軽く振って試料と酸を混和させた後、熱坂上で静かに加熱する。加熱中は、時計皿でふたをする(注3)。
- (c)液量が約15mlになったら、一旦ビーカーを熱板からおろし、硝酸10mlを加えて再び加熱する。硝酸を添加して、加熱するこの操作を二酸化窒素の褐色のガスが発生しなくなるまで繰り返す(注4)。
- (d)ビーカーを熱板からおろし、硝酸5ml、過塩素酸2~3mlを加えて加熱を続け、過塩素酸及び硫酸の白煙を発生させた後(注5)、放冷する。
- (e)ビーカーの壁を少量の水で洗い、再び加熱して硫酸の白煙を十分に発生させ(注6)液量が約5mlになったら放冷する。
- (f)水約50mlを加えて静かに加熱した後、不溶解物が沈降するのをまってろ紙5種Bでろ過する。ビーカー中の不溶解物及びろ紙を水で洗浄する。ろ液及び洗液を合わせ室温まで冷却し全量フラスコ100mlに移し入れ、水を標線まで加え、試験溶液とする。
- 注(2)2.2風乾試料を用いてもよい。
- (3)分解にともなう反応が止んだら時計皿は少しずらすか、ガラス棒を用いるなど適当な方法でうかしておく。
- (4)過塩素酸による有機物の酸化反応は極めて急激で爆発的に進行する。このため、危険のないように硝酸による有機物の酸化を十分に行ってから過塩素酸を添加する。
- (5)過塩素酸白煙が発生したとき、液に着色(黒褐色や褐色)のある場合は直ちに加熱をやめ、放冷後硝酸10mlを加えて再び加熱する操作を繰り返す。
- (6)硝酸が存在すると(4)(d)での水素化ひ素の発生が阻害されるので十分に硫酸の白煙を発生させて硝酸を除去する。
(4)操作
- (a)試験溶液の適量(Asとして0.002~0.01mlを含む)を水素化ひ素発生びんにとり、溶液中にすでに含まれている硫酸との合量が硫酸として約3mlとなるように硫酸(1+1)を加えた後、水で約40mlとする。
- (b)塩酸(1+1)2ml、よう化カリウム(20w/v%)15ml及び塩化すず(Ⅱ)溶液5mlを加えて振り混ぜ、10分間放置する。
- (c)水素化ひ素発生びん、導管及びジエチルジチオカルバミン酸銀溶液5mlを入れた水素化ひ素吸収管を連結した後、水素化ひ素発生びんに亜鉛約5gを手早く投入する。
- (d)水素化ひ素発生びんを約25℃の水浴中に入れ、約1時間放置してジエチルジチオカルバミン酸銀溶液に水素化ひ素を吸収、発色させる。
- (e)この溶液にクロロホルムを加えて正確に5mlとする。
- (f)溶液の一部を吸収セル(10mm)に移し、クロロホルムを対照として波長510nm付近の吸光度を測定する。
- (g)空試験として、ビーカー200ml用い(3)(b)~(f)の操作を行ったものについて(a)~(f)の操作を行い、(f)の吸光度を補正する。
- (h)検量線からひ素量を求める。
- (ⅰ)乾燥試料当りのひ素濃度(mgAs/kg)として結果を表示する。
検量線 ひ素標準液(0.001mgAs/ml)2~10mlを水素化ひ素発生びんに段階的にとり、硫酸(1+1)6mlを加えた後(注7)、水で約40mlとし(b)~(f)の操作を行う。空試験として水約35mlをとり同様の操作を行って標準液について得た吸光度を補正し、ひ素の量と吸光度との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
注(7)鉄を多量に含有する試料を対象とする場合は、これに鉄(Ⅱ)溶液〔塩化鉄(Ⅱ)六水和物5g又は硫酸鉄(Ⅱ)アンモニウム十二水和物9gを塩酸5mlと水に溶かして100mlとする)2mlを加える。
13.2 原子吸光法
(1)試薬
- (a)よう化カリウム溶液(20w/v%)13.1(1)(a)と同じ。
- (b)転化すず(Ⅱ)溶液 転化すず(Ⅱ)二水和物10gを塩酸100mlに溶かす。
- (c)亜鉛粉末JIS K8013〔亜鉛粉末(試薬〕)ひ素分析用。
- (d)鉄(Ⅱ)溶液 塩化鉄(Ⅱ)六水和物5g又は硫酸鉄(Ⅲ)アンモニウム十二水和物9gを塩酸5mlと水に溶かして100mlとする。
- (e)ひ素標準液(0.1μgAs/ml)13.1(1)(g)のひ素標準液(0.001mgAs/ml)10mlを全量フラスコ100mlにとり、塩酸(1+1)2mlを加えた後、水を標線まで加える。
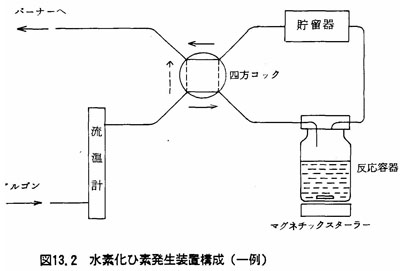
(2)装置
(a)水素化ひ素発生装置 図13.2に示す。
(3)試験溶液の調製
(a)13.1(3)(a)~(f)の操作を行う。
(4)測定操作
- (a)試験溶液の適量(Asとして0.1~1μgを含む)を水素化ひ素発生装置の反応容器にとり、塩酸(1+1)4ml(注8)と水を加えて20mlとする。
- (b)よう化カリウム溶液(20w/v%)2ml、塩化すず(Ⅱ)溶液2ml、鉄(Ⅱ)溶液1mlを加えて振り混ぜ、約15分間放置する。
- (c)水素化ひ素発生装置と原子吸光分析装置を連結し、系内の空気をアルゴン(注9)で置換した後、亜鉛粉末1.0gを手早く(注10)(注11)反応容器中に加え、マグネチックスターラーを作動して水素化ひ素(及び水素)を発生させる(注12)。
- (d)四方コックを回転して、水素化ひ素をアルゴン(注8)-水素フレーム中に導き、波長193.7nmにおける指示値(吸光度又はその比例値)を読む。
- (e)空試験として、200mlビーカーを用い13.1(3)(b)~(f)の操作を行ったものについて(a)~(d)の操作を行って指示値を読み取り、試料について得た結果を補正する。
- (f)検量線からひ素量を求める。
- (g)乾燥試料当りのひ素養度(mgAs/kg)として結果を表示する。
検量線 ひ素標準液(0.1μgAs/ml)1~10mlを反応容器に段階的にとり、塩酸(1+1)4mlを加え、水で20mlとした後、(b)~(d)の操作を行う。空試験として水約15mlをとり同様の操作を行って標準液について得た指示値を補正し、ひ素量と指示値との関係線を作成する。検量線の作成は、試験溶液の測定時に行う。
- 注(8)試験溶液の分取量が多くなり、含有する硫酸量が無視できない場合は、これに対応して塩酸添加量を少くする。
- (9)窒素を用いてもよい。
- (10)水素化ひ素は亜鉛粉末添加直後に急激に発生するのです早く操作を行い、ひ素を逃がさないように注意する。
- (11)亜鉛粉末中には微量のひ素が含まれているので、亜鉛粉末の添加量を一定にする必要がある。このため、1)亜鉛粉末にバインダーを加えて成形した錠剤を添加、2)亜鉛粉末に水を加えて濃い懸濁液としスポイトで添加、3)一定量の亜鉛粉末をオブラートで包んで添加する、などの方法を用いる。また、亜鉛の代わりにテトラヒドロほう酸ナトリウム(水素化ほう素ナトリウム)を還元剤として用いてもよい。この場合は、塩化すず(Ⅱ)溶液及び鉄(Ⅱ)溶液は添加しない。
- (12)発生した気体を貯留する際の補集量の判定には、圧力による方法、体積による方法などがある。
14 シアン化合物
14.1 4-ピリジンカルポン酸-ピラゾロン吸光光度法
(1)試薬
- (a)フェノールフタレイン溶液(0.5w/v%)フェノールフタレイン0.5gをエタノール(95)50mlに溶かし、水を加えて100mlとする。
- (b)アミド硫酸アンモニウム溶液(10w/v%)アミド硫酸アンモニウム(スルファミン酸アンモニウム)10gを水に溶かして100mlとする。
- (c)EDTA溶液(10w/v%)エチレンジアミン四酢酸二ナトリウム二水和物10gを水に溶かし、水酸化ナトリウム溶液(2w/v%)を数滴加えて微アルカリ性とし、水を加えて100mlとする。
- (d)酢酸亜鉛アンモニア溶液(10w/v%)酢酸亜鉛二水和物12gにアンモニア水35mlを加え、水で100mlとする。
- (e)りん酸塩緩衝液(pH7.2) りん酸水素ニナトリウム(無水)17.8gを水約300mlに溶かし、りん酸二水素カリウム溶液(20w/v%)をpH7.2になるまで加え、水で500mlとする。
- (f)クロラミンT溶液(1w/v%)P-トルエンスルホンクロロアミドナトリウム三水和物(クロラミンT三水和物)1.25gを水に溶かして100mlとする。使用時に調製する。
- (g)4-ピリジンカルボン酸-ビラゾロン溶液 トフェニル-3-メチル-5-ビラゾロン0.3gをN,N-ジメチルホルムアミド20mlに溶かす。別に4-ピリジンカルポン酸1.5gを水酸化ナトリウム溶液(4w/v%)約20mlに溶かし、塩酸(1+10)を滴加してpHを約7とする。両液を合わせ、水を加えて100mlとし、冷暗所に保存する。この溶液は10℃以下で保存し、20日間以上経過したものは使用しない。
- (h)N/10硝酸銀溶液、硝酸銀17gを水に溶かし褐色の全量フラスコ1000mlにとり、水を標線まで加える。
標定 塩化ナトリウム(標準試薬)を500~650℃で40~50分間デシケー夕ー中で放冷した後、その1.169gをとり、適量の水に溶かして全量フラスコ200mlに入れ、水を標線まで加える。この20mlをとり、水を加えて液量を約50mlとし、デキストリン溶液(2w/v%)5ml及び指示薬としてフルオレセインナトリウム溶液(0.2w/v%)3~4滴を加え、この硝酸銀溶液で滴定し、黄緑の蛍光が消え、わずかに赤みを呈するときを終点とする。
これに要したN/10硝酸銀溶液の加数(X)から、次式によってファクター(f)を算出する。

- (i)シアン化物イオン標準液(1mgCN-/ml) シアン化カリウム2.51gを水に溶かして1㍑とする。この溶液は使用時に調製する。濃度は、次の方法で求める。
この溶液100mlをとり、水酸化ナトリウム溶液(2w/v%)1mlと、指示薬としてP-ジメチルアミノベンジリデンローダニン〔5-(4-ジメチルアミノベンジリデン)-2-チオキソ-4一チアゾリジノン〕のアセトン溶液(0.02w/v%)0.5mlを加え、N/10硝酸銀溶液で滴定し、溶液の色が黄から赤になったときを終点とする。滴定に要したN/10硝酸銀溶液のml数(a)から、次式によってこの溶液1ml中のシアン化物イオン(CN-)の量(mg)算出する。

- (j)シアン化物イオン標準液(0.001mg CN-/ml) シアン化物イオン標準液(1mg CN-/ml)の10mlを全量フラスコ1000mlに入れ、水酸化ナトリウム溶液(2w/v%)100mlにとり、水を標線まで加える。使用時に調製する。溶液の濃度は、シアン化物イオン標準液(mg CN-/ml)の濃度から算出する。
(2)装置
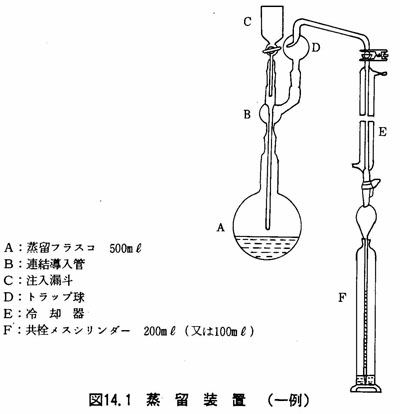
(a)蒸留装置 図14.1に示す。
(3)試験溶液の調製
- (a)2.1の湿試料5~10g(注1)を0.1gのけたまで蒸留フラスコ500mlにはかり取り、水250mlを加える。
- (b)指示薬としてフェノールフタレイン溶液(0.5w/v%)数滴を加える。アルカリ性の場合は、溶液の赤い色が消えるまでりん酸で中和する。
- (c)次にアミド硫酸アンモニウム溶液(10w/v%)1mlを加える(注2)。
- (d)蒸留フラスコを接続し、受器には共栓メスシリンダー200mlを用い、これに水酸化ナトリウム溶液(2w/v%)20mlを入れ、冷却管の先端を受液中に浸す。
- (e)蒸留フラスコにりん酸10mlを加え、次にEDTA溶液(10w/v%)20ml(注3)を加える。
- (f)数分間放置した後、蒸留フラスコを加熱し、留出速度2~3ml/minで蒸留する(注4)(注5)。受器の液量が約150mlになったら、冷却管の先端を内容液から離して蒸留を止める。冷却管の内外を少量の水で洗い、洗液は留出液と合せる。
- (g)酢酸亜鉛アンモニア溶液(10w/v%)10ml(注6)を留出液に加え、よく振り混ぜた後、水を標線まで加えて振り混ぜ、約30分間放置する(注7)。
- (h)ろ紙5種Cでろ過する。
- (i)ろ液100mlを蒸留フラスコ500mlにとり、水150mlを加え、指示薬としてフェノールフタレイン溶液(0.5w/v%)数滴を加え、りん酸で中和する。
- (j)受器として共栓メスシリンダー100ml用い、(d)と同様に操作する。
- (k)(e)と同様に操作する。
- (l)(f)と同様に操作し、留出液が約90mlになるまで蒸留する。
- (m)水を標線まで加えて振り混ぜ、これを試験溶液とする。
- 注(1)水質試験では、採取後直ちにアルカリ性として、シアン化合物を固定するが、底質試料に同様の処理を行うと、シアン化合物が液相に移行し損失する恐れがあるので、現地処理をせず、試料は0~10℃の冷蔵庫に保存し、できるだけ速やかに試験する。
- (2)亜硝酸イオンが共存すると、EDTAと反応してシアン化合物が生成する。アミド硫酸アンモニウム溶液は亜硝酸イオンの妨害を除くために添加する。10w/v%溶液1mlは亜硝酸イオン約35mgに対応する。
- (3)底質中のシアン化合物は、多くの金属イオンと反応して金属錯体として存在している場合が多い。EDTAはこのような金属イオンをマスキングする。金属イオンが多い場合はEDTA溶液の添加量を増やす。
- (4)留出速度が早いとシアン化水素が完全に留出しないので、3ml/min以上にしない。
- (5)蒸留中、冷却管の先端は常に液面下15mmに保つようにする。
- (6)留出液中に流化物イオンが共存すると、ピリジンービラゾロン法等の吸光光度法で負の誤差を生じるので、酢酸亜鉛アンモニア溶液を加えて沈殿除去する。この溶液(10w/v%(1mlは硫化物イオン約14mgに相当する。
- (7)硫化亜鉛の白色沈殿と共に、水酸化亜鉛の白色沈殿が生じる。水酸化亜鉛の沈殿生成により溶液のpHが低下し、更に沈殿が生成するので、しばらく放置する。
(4)操作
- (a)(3)で調製した試験溶液の適量(25ml以下、CN-として0.5~9μgを含む)を全量フラスコ50mlにとる。
- (b)指示薬としてフェノールフタレイン溶液(0.5w/v%)1滴を加え、静かに振り混ぜながら酢酸(1+8)を滴加して中和した後、りん酸塩緩衝液(pH7.2)10ml(注8)を加え、密栓して静かに振り混ぜる。
- (c)クロラミンT溶液(1w/v%)0.5mlを加え、直ちに密栓して静かに振り混ぜ、約5分間放置する。
- (d)4-ピリジンカルポン酸-ビラゾロン溶液10mlβを加え、更に水を標線まで加え、密栓して静かに振り混ぜた後25±2℃の水浴中に30分間放置する(注9)。
- (e)溶液の一部を吸収セルに移し、波長638nm付近の吸光度を測定する。
- (f)空試験として水10mlを全量フラスコ50mlにとり、りん酸塩緩衝液(pH7.2)10mlを加えた後、(c)~(e)の操作を行い、(e)の吸光度を補正する。
- (g)検量線からシアン化物イオンの量を求め、別に3乾燥減量で求めた乾燥減量(%)を用いて、乾燥試料当りの濃度(mg CN-/kg)として結果を表示する。
検量線 シアン化物イオン標準液(0.001mg CN-/kg)0.5~9mlを全量フラスコ50mlに段階的にとり、液量を約10mlとして、(b)~(f)の操作を行って、シアン化物イオン(CN-)の量と吸光度との関係線を作成する。
- 注(8)発色時のpHは7~8の範囲に入らなければならない。
- (9)20℃以下では、十分に発色せず、また30℃以上では発色も早いが退色も早くなる。
14.2 ピリジン-ビラゾロン吸光光度法
(1)試薬
- (a)フェノールフタレイン溶液(0.5w/v%)14.1(1)(a)と同じ。
- (b)アミド硫酸アンモニウム溶液(10w/v%)14.1(1)(b)と同じ。
- (c)EDTA溶液(10w/v%)14.1(1)(c)と同じ。
- (d)酢酸亜鉛アンモニア溶液(10w/v%)14.1(1)(d)と同じ。
- (e)りん酸塩緩衝液(pH6.8) りん酸二水素カリウム17.0gとりん酸水素ニナトリウム(無水)17.8gを水に溶かして500mlとする。
- (f)クロラミンT溶液(1w/v%)14.1(1)(f)と同じ。
- (g)ピリジン-ビラゾロン溶液 トフェニル-3-メチルー5-ビラゾロン0.25gを75℃の温水100mlに溶かして室温まで冷却する(完全に溶けなくても差し支えない)。これにビス(1-フェニルー3-メチル-5-ビラゾロン)0.02gをピリジン20mlに溶かした液を加えて混ぜる。使用時に調製する。
- (h)シアン化物イオン標準液(0.001mg CN-/ml)14.1(1)(j)と同じ。
(2)装置
(a)蒸留装置 14.1(2)(a)と同じ。
(3)試験溶液の調製
14.1(3)(a)~(m)操作を行い、試験溶液を調製する。
(4)操作
- (a)(3)で調製した試験溶液の適量(25ml以下、CN-として0.5~9μgを含む。)を全量フラスコ50mlにとる。
- (b)指示薬としてフェノールフタレイン溶液(0.5w/v%)を1滴加え、静かに振り混ぜながら溶液の赤い色が消えるまで酢酸(1+8)を滴加する。
- (c)りん酸塩緩衝液(pH6.8)10mlを加え(注10)、密栓して静かに振り混ぜる。
- (d)これにクロラミンT溶液(1w/v%)0.25mlを加え、直ちに密栓して静かに振り混ぜ約5分間放置する。
- (e)ピリジンービラゾロン溶液15mlを加え、更に、水を標線まで加え、密栓して静かに振り混ぜる。
- (f)25±2℃の水溶中に約30分間(注11)浸し、溶液の色が薄い紅から紫を経て安定な青になるまで発色させる。
- (g)溶液の一部を吸収セルに拶し、波長620nm付近の吸光度を測定する。
- (h)空試験として水10mlを全量フラスコ50mlにとり、(c)~(g)の操作を行い、(g)の吸光度を補正する。
- (i)検量線からシアン化物イオンの量を求め、別に3乾燥減量で求めた乾燥減量(%)を用いて、乾燥試料当りの濃度(mg CN-/kg)として結果を表示する。
検量線 シアン化物イオン標準液(0.001mg CN-/ml)0.5~9mlを全量フラスコ50mlに段階的にとり、水を加えて約10mlとし、(b)~(h)の操作を行って、シアン化物イオン(CN-)の量と吸光度との関係線を作成する。
- 注(10)前処理して得られたシアン化物イオン溶液のpHは約13になっており、この溶液10mlを中和するのに必要な酢酸(1+8)は約0.5mlで、これにりん酸塩緩衝液(pH6.8)10mlを加えるとpH6.8になる。発色時のpHは5~8の範囲に入らなければならない。
- (11)20℃以下では、十分に発色せず、また30℃以上では発色も早いが退色も早くなる。
15 ポリ塩素化ビフェニル(PCB)
試料のアルカリ分解処理を行い、PCB以外の油脂分や有機化合物等をけん化処理してから、PCBその他未分解の有機化合物をヘキサンで抽出し、シリカゲルカラムクロマト管により、妨害物質除去操作を行った後、ヘキサン浮出液について、ガスクロマトグラフによりPCBを測定する方法である。
(1)試薬
- (a)水 水1㍑につきヘキサン100mlを加えて振り混ぜ洗浄したもの。
- (b)ヘキサン ヘキサン300mlを濃縮器を用いて約3mlに濃縮し、その10μ㍑を分取して、ガスクロマトグラフに注入した時、PCBの保持時間にピークを生じないもの(注1)。
- (c)水酸化カリウムのエタノール溶液 水酸化カリウム35gをできるだけ少量の水に溶かし、エタノール(95)500mlを加えて振り混ぜ、二酸化炭素に触れないように、2~3日放置したのち、その上澄み液を取るか、ろ過するかして、耐アルカリ性のびんに保有する。
- (d)硫酸ナトリウム(無水) 共栓三角フラスコに硫酸ナトリウム(無水)100gを取り、これにヘキサン50mlを加え栓をして振り混ぜ、ヘキサンをろ別する。再び残分にヘキサン25mlを加えて振り混ぜ、ヘキサンをろ別する。この硫酸ナトリウムを取り、風乾したものを用いる。この場合、後段でろ別したヘキサンの10μ㍑を分取してガスクロマトグラフに注入したとき、PCBの保持時間にピークを生じないこと。
- (e)シリカゲル PCB分析用シリカゲル粉末をビーカーに入れ、層の厚さを10mm以下にして130℃で約18時間乾燥した後、デシケータ中で約30分間放冷し、直ちに使用する。
- (f)PCB混合標準液 試験用PCBのKC-300、KC-400、KC-500、及びKC-600(注2)を重量比1:1:1:1の割合で混合したものをヘキサンに溶かし、0.01~1mg/㍑の濃度となるように調製する。
- (g)シリカゲル分画試験液 KC-300のヘキサン溶液(0.001w/v%)とKC-600のヘキサン溶液(0.001w/v%)を、容量比2対1の割合で混合する。
- 注(1)一般にヘキサンを70℃付近で蒸留し、その中留をとる。また、市販の残留農薬分析用試薬を用いることができる。
- (2)試験用三塩素化ビフェニル及び試験用六塩素化ビフェニルなどは、一般にKC-300及びKC-600などの名称で入手できる。
(2)器具及び装置(注3)
- (a)還流冷却器 例を図15.1に示す。
- (b)フラスコ 容量200mlですり合わせ共栓付きのもの。
- (c)濃縮器 クデルナダニッシュ濃縮器(以下「KD濃縮器」という。例を図15.2に示す。)又は、ロータリーエバボレーター。
- (d)カラムクロマトグラフ用ガラス管(以下「クロマト管」という)
内径約10mm、長さ約300mmのコック付きガラス管(例を図15.3に示す)。 - (e)マイクロシリンジ 容量1~10μ㍑のもの。
- (f)ガスクロマトグラフ
試料導入部 温度を200~250℃にしたもの。
分離管 内径2~4mm、長さ150~200㎝のガラス製のものであって、その温度を180~250℃(トリチウムを用いた検出器を使用する場合は、180~220℃)にしたもの。
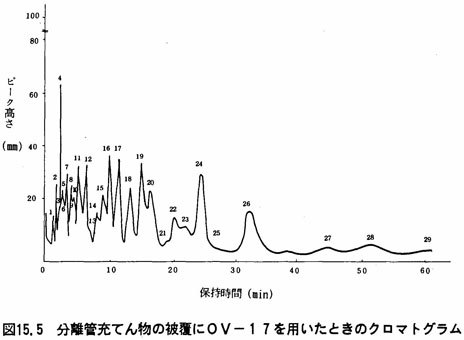
分離管充てん物 酸で洗浄した後シラン処理をしたガスクロムQ、クロモソルプG又はクロモソルプW(いずれも粒径149 ~177μmのもの)に0V-1又は、0V-17を1.5~5%被覆したもの。
検出器 電子捕獲検出器。その温度を200~250℃(トリチウムを使用する場合は、200~220℃)にしたもの。
キャリヤーガス 99.9v/v%以上の窒素又はヘリウムであって、流量を毎分30~80mlとしたもの。
注(3)ガラス器具類については、あらかじめヘキサンで洗浄し、乾燥したものを用いる。すり合わせ部にワセリン等を使用してはならない。
(3)試験溶液の調製
(3).1.1試料のアルカリ分解
備考 例図のKD濃縮器を用いて蒸留濃縮を行うとき、スナイター管では蒸留及び還流が各段のスナイダー球のふし目で繰り返して行われるため、溶媒の蒸留に際してPCBの損失が殆んど無く、濃縮効果が大きい。
このとき、蒸留器の系内を減圧にすると、溶媒の沸点が下がり蒸留速度は早まるが、各すり合わせ部分から外気が吸引され、大気中の異物が混入する危険があるため、減圧にはしない。
突沸を避けるには、加熱部の濃縮管内に毛細管等の小片を入れておくとよい。外気を少しずつ入れて気泡を出させるためのキャピラリー管は取り外し外気の混入を老ける。
蒸留操作中、スナイダー管での蒸留が緩慢なときには、この部分をアルミホイル等で覆うと温度が上昇し留出が早くなる。
ロータリーエバボレーターを使用した場合も外気からの異物の混入に注意する。
- (a)2.1の湿試料約10gを0.1gのけたまではかり取り、これを還流冷却器付フラスコに入れ、水酸化カリウムのエタノール溶液50mlを加え約1時間沸騰水浴上で加熱分解を行い約50℃まで放冷する(注4)。
- (b)フラスコにヘキサン50mlを加えて激しく振り混ぜ静置して室温まで放冷した後、ガラス繊維を敷いた漏斗(注5)を用いてろ過し分液漏斗に入れる。フラスコの内容物はヘキサン20mlずつで3回洗い洗液をろ過して分液漏斗に合わせる。
- (c)分液漏斗に水の総量が約50mlになるように水を追加し、緩やかに振り混ぜた後ヘキサンが十分分離するまで静置し(8)、ヘキサン層を分離し、水層を他の分液漏斗に入れ再びヘキサン50mlを加え同様に抽出を行い、ヘキサン層を先のヘキサン層と合わせる。更にヘキサン層を水100mlずつで緩やかに振り混ぜ3回洗浄する。
- (d)洗浄したヘキサン層を硫酸ナトリウム(無水)約10gの分離管に通して濃縮器で受けた後、水浴上で、液量が約5mlになるまで濃縮する。
- 注(4)温度が高いときにヘキサンを加えると、沸騰するので危険である。また、あまり冷却しすぎるとアルカリ分解によって生成したけん化物がヘキサンに溶けにくくなり、PCB抽出の低下の原因となる。
- (5)ガラス繊維ろ紙を用いてもよいが、目づまりを起こしやすくろ過が困難になることが多い。
- (6)エマルジョンを生ずる場合は、エタノール数mlを加え、穏やかに振り混ぜる。
(3).1.2 アセトニトリル・ヘキサン分配法による油分等の除去操作(注7)
注(7)油分等を多く含む底賓で、(3).1.1の操作によっても分解されず、多量に油分等の残る試料については、次の「(3).2 シリカゲルカラムクロマト管による妨害物質除去操作」を行う前に、この操作を行う。
- (a)(3).1.1の操作により得た濃縮液を少量のヘキサンを用いて100mlの分液漏斗に洗い込み(総ヘキサン量は約15mlとする)、ヘキサン飽和アセトニトリル(ヘキサン約30mlとアセトニトリル約100mlを分液漏斗に入れ振り混ぜた後アセトニトリル層を使用する)30mlを加え約1分間激しく振り混ぜ抽出した後、静置する。
- (b)アセトニトリル層を1000mlの分液漏斗へ移し入れ、ヘキサン層をヘキサン飽和アセトニトリル30mlずつで更に3回抽出繰作を行う。
- (c)アセトニトリル層を合わせ、塩化ナトリウム溶液(2%)700mlとヘキサン100mlを加え、穏やかに振り混ぜ十分分離するまで静置する。
- (d)水層を別の分液晶斗に入れ、これにヘキサン100mlを加え、激しく振り混ぜ、静置した後、水層を捨て、先のヘキサン層と合わせ、更に塩化ナトリウム溶液(2%)100mlずつで2回洗浄する。
- (e)ヘキサン層に硫酸ナトリウム10gを加えて振り混ぜ、水分を除いてから、ヘキサン層を濃縮器に移し入れ、約5mlまで濃縮する。
(3).2 シリカゲルカラムクロマト管による妨害物質除去操作(注8)
注(8)この操作を行うにあたり、シリカゲルの確認及びポリ塩素化ビフェニルの流出範囲を調べるため、シリカゲルカラムクロマト管溶出試験を行った後、妨害物質除去操作を行う。
(3).2.1 シリカゲルクロマト管溶出試験
(1)シリカゲルカラムの作り方
- (a)クロマト管の底部に脱脂綿又はガラス繊維を詰め、へキサン10mlでクロマト管内を洗い、脱脂綿又はガラス繊維上部までヘキサンを残す。
- (b)シリカゲル2gをヘキサン10mlを入れたビーカーにはかりとり、ガラス棒でゆるやかにかき混ぜて気泡を除き、クロマト管に充てんする。
- (c)ヘキサンを流下させ、シリカゲル層を安定させた後、硫酸ナトリウム(無水)1gをシリカゲルの上にのせる。
- (d)ヘキサン2mlを駒込ピペットを用いて、クロマト管内壁に付着している硫酸ナトリウム(無水)を洗い落とした後、シリカゲル分画試験液2mlをピペットで静かに硫酸ナトリウム(無水)の上に加え、下端のコックを開いて溶液を硫酸ナトリウム(無水)の面まで下げる。
- (e)クロマト管内壁をヘキサン1mlで洗い、液面を硫酸ナトリウム(無水)の面まで下げる。更にヘキサン1mlでクロマト管内壁を洗い、洗液は、硫酸ナトリウム(無水)の面まで下げる。
(2)PCBの流出範囲の測定
- (a)(1)で作ったシリカゲルカラムをクロマト管の上に、分液漏斗300mlを付け、ヘキサン200mlを加え、下端のコックを開いて、毎秒1滴の割合でヘキサンを流下させる。
- (b)流出液は、10mlごとに集め、試験管20本に分取する。それぞれの流出画分について5~10μ㍑をガスクロマトグラフに注入し、ポリ盤素化ビフェニルの流出開始と終了点を測定する。
- (c)この操作を2~3回繰り返し、流出範囲の安定性と回収率を十分に確認しておく。別に、シリカゲル分画試験液を用いないでこの操作を行い、そのときの測定条件下のガスクロマトグラフで妨害ピークの有無を調べておき、数値化の段階で考慮する。
(3).2.2 妨害物質除去操作
- (a)(3).2.1(1)で作ったクロマト管の上に、(3).1の操作により得た濃縮液を移し入れ、コックを操作して溶液を硫酸ナトリウム(無水)面まで下げる。濃縮液の入っていた容器をヘキサン約2mlずつで数回洗い、洗液を濃縮液に静かに合わせる。更にクロマト管内壁を少量のヘキサンで洗った後、液面を硫酸ナトリウム(無水)面まで下げる。
- (b)クロマト管にヘキサンを入れた分液漏斗300mlを付け、下端のコックを開けて、溶液を(3).2.1(2)で求めたPCB流出範囲まで流出する。
- (c)KD濃縮器にガラス毛細管数本を入れ、水浴上で液量が5mlになるまで濃縮し、これを試験溶液とする。
(4) 操作
(4).1 ガスクロマトグラフによるPCBの測定
- (a)適当な濃度のPCB混合標準液5μ㍑をマイクロシリンジを用いてガスクロマトグラフに注入する。
- (b)得られたクロマトグラムのパターンについて図15.4又は図15.5を参考にしてピーク番号ごとにピーク高さ(mm)を涜み取る。
- (c)(3).2.2で得た試験溶液の適量(1~10μ㍑)をマイクロシリンジを用いてPCB混合標準液と同一条件でガスクロマトグラフに注入する。
- (d)得られたクロマトグラムのパターンについてピーク番号ごとにピーク高さ(mm)を読み取る。
(4).2 測定値の算出
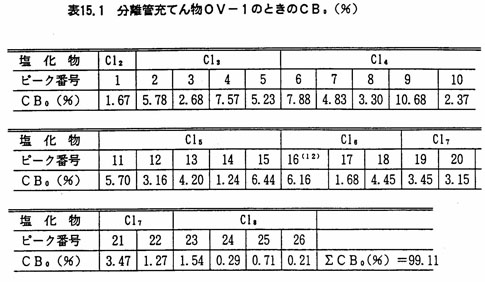
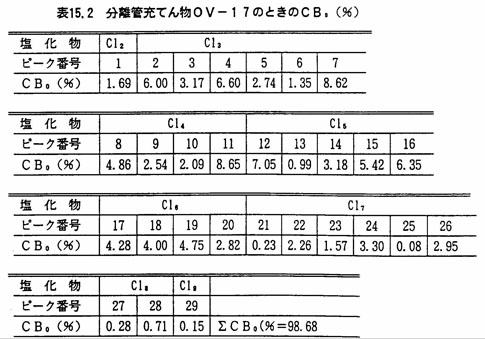
PCB量を、次の方法によって算出する。CB0(%)(注9)として表15.1又は表15.2に示したものを用いる。
- (a)(4).1.(b)で読んだ各ピーク番号ごとのピーク高さ(H1mm)を求め、K値く10)を次式で算出する。
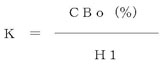
- (b)(4).1.(d)で読んだ各ピーク番号ごとのピーク高さ(H2mm)から試験溶液中のCB2(%)を求める。
CB2(%)=K×H2
- (c)試料中のPCB量(mgPCB/kg)を、次の式によって算出する。なお、次式により、

- 注(9)電子捕獲検出器の相対感度が条件により変動が大きいことから、各種PCB混合標準液を用い、図15.4又は図15.5のピーク番号、及び表15.1又は表15.2の各ピーク含有率から検量線を作成し、あらかじめ直線性のある測定範囲を定めておく。ここでCBo(%)は、PCB混合標準液を、ガスクロマトグラフ-質量分析計を用いて各ピークの塩素原子数を明らかにし、ガスクロマトグラフ(水素イオン化検出器)のクロマトグラムからPCBの各ピークごとの成分割合を求めたものである。
- (10)qK値は線源などのガスクロマトグラフ操作条件が異なれば変動する。したがって試料の測定にあたり、必ずそれと同一条件でPCB混合標準液のガスクロマトグラムからK値を算出する。
- (11)3.2で求めた試料の乾燥減量(%)から乾燥試料の質量を求める。
- (12)ピーク番号16は条件により、16(CBo(%)2.16)及び16(CBo(%)4.00)に分離することがある。
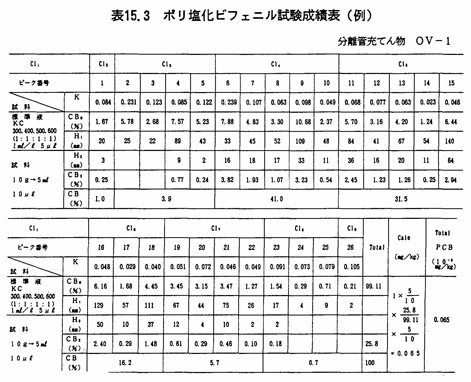
上の成績表(例)は、試料10gを用いて、本文(3).2で得た溶液から10μ㍑をとり、ガスクロマトグラフに注入して測定した場合の例である。この時のPCB混合標準液の濃度は1mg/㍑、注入量5μ㍑(5ng)である。
これによると、試料中のPCB濃度は次のように求められる。
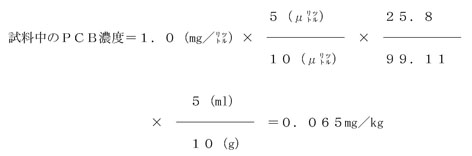
なお、PCB混合標準液のピーク番号5について見るとき、ピーク高さは43mm、そのPCB含有率は5.23%であるから、このピークのPCB量は
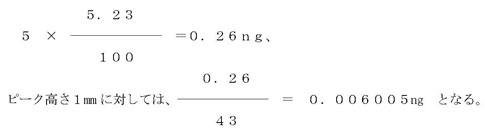
16 ヘキサクロロシクロヘキサン(HCH、旧称BHC)
試料中のHCHをアセトンで抽出した後、ヘキサンに転溶させ、フロリジルカラムにより妨害物質除去操作を行い、ガスクロマトグラフにより測定する。
(1)試薬
- (a)フロリジル アルミナ製又は磁製の皿に入れ、130℃で15時間加熱する。加熱後デシケーター内で放冷する。フロリジルの性能は、ロット毎に若干変動するのであらかじめHCHの溶出範囲を調べておく。
- (b)水 水1㍑につきヘキサン100mlを加えて振り混ぜ洗浄したもの。
ヘキサン(注1)試薬特級品を70℃付近で蒸留し、その中留をとる。
アセトン(注2)試薬特級品を蒸留精製したもの。
HCH標準液(100mg/㍑)HCHの4異性体(α、β、γ、σ)の各標準品100mgを全量フラスコ100mlにはかりとり、少量のベンゼン又はトルエンを用いて溶かし、ヘキサンを加えて100mlの溶液とする。
この溶液10mlを全量ピペットを用いて全量フラスコ100mlにとり、ヘキサンを加えて100mlの溶液とする。 - (f)HCH標準液(注3)HCH標準液(100mg/㍑)をヘキサンを用いて希釈し、適当な濃度(0.01mg/㍑~0.1mg/㍑)の溶液を調製する。
なお、4異性体を混合して調製してもよい。 - (g)塩化ナトリウム溶液(2w/v%)
- (f)硫酸ナトリウム(無水)硫酸ナトリウム(無水)100gにヘキサン50mlを加えて振り混ぜ、ろ別する。残分にヘキサン25mlを加えて混ぜ、ろ過したのち風乾する。この場合、ろ別したヘキサンの10μ㍑を分取してガスクロマトグラフに注入したとき、HCHの保持時間にピークを生じないものを用いる。
- (i)銅チップ 直径1mm程度の銅線を2~3mmの長さに切断したもの(10~20g)を用意しガラス共栓試験管(25~30mlに入れ濃塩酸15mlを加えて激しく振り混ぜる。
傾斜して塩酸を除去した後、蒸留水を加えて水洗し、塩酸を十分除去した後、アセトンで洗い水分を除き、ドライヤー等を用いて手早く乾燥する。
この銅のチップは、ヘキサン中に保存する(使用後表面の黒化した銅チップもこの方法で再生する。)。 - (j)エチルエーテル含有ヘキサン溶液 ヘキサンに容量比率6%になるようにエチルエーテルを加え混合する。
- 注(1)(2)市販の「残留農薬用試薬」を用いることができる。
- (3)市販のHCH標準液を用いて調製することができる。
(2)器具及び装置(注4)
- (a)フラスコ15.(2)(b)と同じ。
- (b)濃縮器15.(2)(c)と同じ。
- (c)クロマト管15.(2)(d)と同じ。
- (d)マイクロシリンジ 容量1~10μ㍑のもの。
- (e)ガスクロマトグラフ
試料導入部 温度を180~250℃にしたもの。
分離管 内径3~4mm、長さ100~200㎝のガラス製のものであって、その温度を150~200℃にしたもの。
分離管充てん物 酸で洗浄した後シラン処理をしたガスクロムQ、クロモソルブG又はクロモソルプW(いずれも粒径149~177μmのもの)に0V-17を5%被覆したもの。
検出器 電子捕獲検出器。その温度を180~250℃(トリチウムを使用する場合は、180~220℃)にしたもの。
キャリヤーガス 99.9v/v%以上の窒素又はヘリウムであって、流量を毎分50~100mlとしたもの。
注(4)ガラス器具類については、あらかじめヘキサンで洗浄し、乾燥したものを用いる。すり合わせ部にワセリン等を使用してはならない。
(3) 試験溶液の調製
(3).1 抽出操作
- (a)2.1の湿試料薬20gを0.1gのけたまで共栓付フラスコにはかり取り、アセトン50mlを加え20分間激しく振り混ぜる。振り混ぜた後しばらく放置して、ろ紙5種Bでろ過する。ろ紙上の残さを少量の水とアセトン50mlを用いてフラスコ内に洗い入れる。再度30分間激しく振り混ぜ、ろ過する。
- (b)ろ液を合わせて役30mlまで濃縮器を用いて濃縮する。
- (c)濃縮液に転化ナトリウム溶液(2w/v%)300ml、ヘキサン100mlを加え、5分間激しく振り混ぜる。ヘキサン層を分離し水層に再びヘキサン100mlを加え5分間激しく振り混ぜる。ヘキサン層を分離し、先のヘキサン層と合わせ100mlの水で2回洗浄する。
- (d)無水硫酸ナトリウムを用いて脱水し、ヘキサン層を傾斜法又はろ紙5種Bでろ過する。少量のヘキサンで無水硫酸ナトリウムを洗浄し、ろ液と洗液を合わせる。
- (e)ヘキサン層をKD濃縮器を用いて役5mlまで濃縮し、フロリジルカラムクロマト用試料とする。
(3).2 フロリジルカラムクロマト管による妨害物質除去操作(注5)
- (a)クロマト管の底部に脱脂綿又はガラス繊維を詰めヘキサンで洗浄する。
- (b)フロリジル5gにヘキサンを加えてかき混ぜ、気泡を除きカラムに充てんしヘキサン10mlを流下させる。無水硫酸ナトリウム1gを上積した後、(3).1で得た濃縮液を静かに流下させ少量のヘキサンを用いてガラス壁を洗う。
- (c)エチルエーテル含有ヘキサン溶液100mlを用いて溶出を行い、これを濃縮して一定容量とし、試験溶液とする。
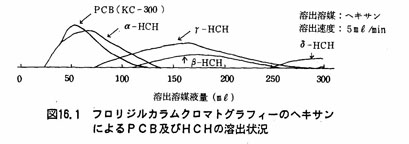
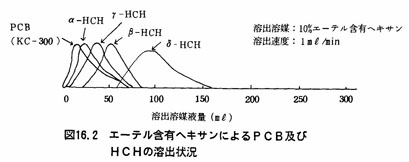
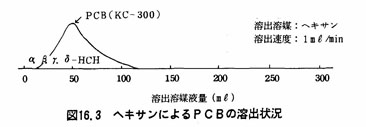
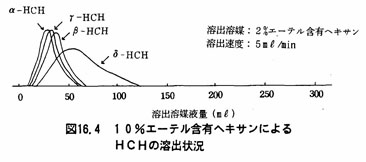
注(5)フロリジルカラム法は、試験溶液中の着色物質等を除く操作である。フロリジルカラムクロマトグラフィーによるPCBおよびHCHの溶出の状況は図16.1及び16.2に示すとおりである。図16.1からわかるように溶出媒体としてヘキサンを用いた場合、PCB、α-HCHのみは溶出溶媒液量200mβで溶出されるが、β-HCH、γ-HCH及びσ-HCHは完全には浮出されない。しかし、図16.2のように2%エーテル含有ヘキサンを用いて溶出を行うと溶出溶媒液量200mlでPCB、α一HCH、β-HCH、γ-HCHは完全に溶出する。
(3).3 シリカゲルカラムクロマト管によるPCBの分離(注6)
注(6)PCBが共存する場合は、(4)のガスクロマトグラフにより測定を行ったとき、クロマトグラム上で、PCBの一部とHCHが重なることがある。この場合、PCBの項の15.(3).2妨害物質除去操作に従って、PCBを分離して測定する。
この方法は、ヘキサンで初めにPCBを溶出した後、HCHを10%エチルエーテル含有ヘキサンで溶出する。
- (a)15(3)2.1によって作ったクロマト管の上に(3).2の操作で得た試験溶液を移し入れ、15(3).2.2の操作に従ってPC8画分を流出する。
- (b)ヘキサン450mlにエチルエーテル50mlの割合で混合した溶液を分液漏斗300mlに入れ、クロマト管に取り付け、下端のコックを開けて、HCH流出範囲(注7)まで流出する。
- (c)流出液を合わせ、濃縮器を用いて濃縮し、一定容量とし試験溶液とする。
注(7)HCHの流出範囲は、あらかじめPCB標準液及びHCH標準液の混合溶液を用いて(a)以後と同様に操作し、各成分の流出範囲を求めておく。図16.3及び図16.4にその例を示した。例では、ヘキサンで溶出を行うと初めPCBだけが溶出し、HCHはシリカゲルカラムに残留する。次に10%エーテル含有ヘキサンでHCHが溶出する。
(3).4 銅ちっぷ処理(注8)
注(8)分子状硫黄が共存し、ガスクロマトグラフによる定量を妨害する場合は、あらかじめ銅チップ処理を行う。
- (a)(3).2(c)で得た試験溶液を共栓付ガラス試験管に取り銅チップ約5gを加えて約30秒間激しく振り混ぜる。
- (b)しばらく静置後、上澄み液を駒込ピペットで注意深く別の共栓試験管に移し、再び数粒の銅チップを加え、軽く振り混ぜる。これで銅チップが黒変しなければガスクロマトグラフ測定に供する。
黒変した場合、再び5g程度の銅チップを加えて激しく振り混ぜる。
以上同様にして銅チップの表面に黒変がなくなるまで反復処理する。
(4)操作
(4).1 ガスクロマトグラフによるHCHの測定
- (a)HCHの標準溶液を段階的にガスクロマトグラフに注入し、ガスクロマトグラムを得、HCHの4異性体の量と各ピーク面積と関係線を作成する。
- (b)(3).2で得た試験溶液の適量(1~10μ㍑)をマイクロシリンジを用いてガスクロマトグラフに注入する。
- (c)得られたガスクロマトグラムについて、HCHの4異性体の各ピーク面積を測定し(9)、あらかじめ作成した検量線からHCH量を求める。
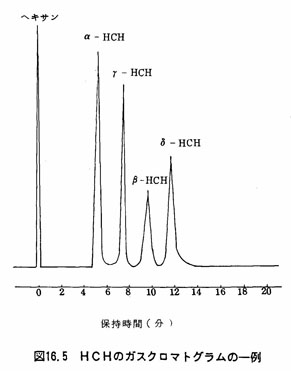
注(9)図16.5にHCHのガスクロマトグラムの一例を示す。
(4).2 測定値の算出

注(10)3乾燥減量で求めた乾燥減量(%)を用いて、乾燥試料当りの質量を求める。
17 硫化物
(1)試薬
- (a)亜鉛アンミン溶液 硫酸亜鉛七水和物5gを水約500mlに溶かし、これに水酸化ナトリウム6gを水約300ml溶かした溶液を加える。次いで硫酸アンモニウム70gをかさ混ぜながら加え、水酸化亜鉛の沈殿を完全に溶かし、水を加えて1㍑とする。
- (b)酢酸亜鉛溶液(10w/v%) 酢酸亜鉛二水和物12gを水に溶かして100mlとする。
- (c)よう素溶液(N/100) よう素1.27gをよう化カリウム5gとともに、約50mlの水に溶かし、水を加えて1㍑とする。
- (d)N/10チオ硫酸ナトリウム溶液 チオ硫酸ナトリウム五水和物26g及び炭酸ナトリウム(無水)0.2gをとり、水に溶かして1㍑とし、これに3-メチル-1-ブタノール(イソアミルアルコール)約10mlを加え、よく振り混ぜて2日間放置する。
N/100チオ硫酸ナトリウム溶液 N/10チオ硫酸ナトリウム溶液20mlを全量フラスコ200mlにとり、水を標線まで加える。この溶液は使用時に調製する。ファクターは、N/10チオ硫酸ナトリウム溶液のものを用いる。
標定 よう素酸カリウム(標準試薬)を120~140℃で2時間乾燥し、デシケーター中で放冷した後0.7134gをとり、水に溶かし、全量フラスコ200mlに入れ、水を標線まで加える。この20mlをとり、共栓三角フラスコ300mlに入れ、よう化カリウム2g及び硫酸(1+5)5mlを加え、直ちに栓をして静かに振り混ぜ、暗所に5分間放置する。水100mlを加え、遊離したよう素をN/10チオ硫酸ナトリウム溶液で滴定する。溶液の黄色が薄くなったら、指示薬としてでんぶん溶液1mlを加え、よう素でんぶんの青い色が消えるまで滴定する。
別に、同一条件で空試験を行って補正したml数から次式によってN/10チオ硫酸ナトリウム溶液のファクター(f)を算出する。

- (e)でんぶん溶液 でんぶん(溶性)1gを水約10mlに混ぜ、熱水100ml中に、かき混ぜながら加え、約1分間煮沸した後、放冷する。使用時に調製する。
(2)装置
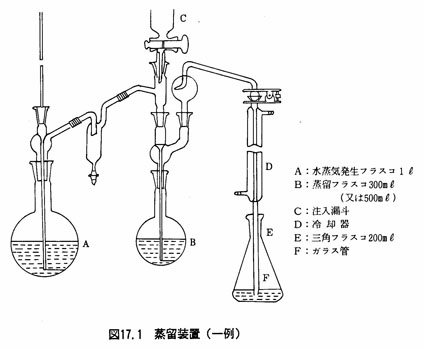
図17.1に示すような蒸留装置を用いる。冷却器の管の先端には、先を細長く引いたガラス管をゴム管で連結し、交換できるようにする。水蒸気発生フラスコは丸底フラスコ1㍑、蒸留フラスコは丸底フラスコ300~500ml、受器は三角フラスコ200mlを用いる。
(3)採泥時の試料の調製(注1)
- (a)試料採取に先立って、ポリエチレンびん300mlに亜鉛アンミン溶液を満たしておく。
- (b)採取した試料を均一に混ぜ、約50gを(a)のポリエチレンびんにとり、亜鉛アンミン溶液をあふれさせ、容器中に空気が残らないように密栓してよく混和した後、5℃以下に保有する。
注(1)硫化物は不安定で空気にさらされると揮散したり酸化分解されたりするので、亜鉛アンミン溶液を加え、現地で固定する。この保存方法をとった場合でも、できるだけ速やかに試験を行う。
(4)試験溶液の調製
- (a)(3)の試料をよく混合した後、その一部を孔径約1μmのガラス繊維ろ紙(注2)を用いて手早く吸引ろ過し、ろ紙上の残留物の適量(注3)を0.01gのけたまで蒸留フラスコ300~500mlにはかり取るく(注4)(注5)。
- (b)別にろ紙上の残留物について3乾燥減量により乾燥減量(%)を測定する。
- (c)(a)の蒸留フラスコ300~500mlに水20~30mlを加えて混和する。
- (d)受器に酢酸亜鉛溶液(10w/v%)20mlを入れ、ガラス管の先端を受液中に浸す。
- (e)注入用漏斗から硫酸(1+5)5mlを加えた後、蒸留フラスコを加熱し、沸騰し始めたら水蒸気を蒸留フラスコに送って水蒸気蒸留を行う。
- (f)受器の内容液が約100mlになったら、ガラス管の先端を内容液から離して蒸留を止める(注6)。
- (g)ガラス管をはずして受器に入れる(注7)。
これを試験溶液とする。 - (h)別に水30mlを用いて(c)~(g)の繰作を行う。これを空試験溶液とする。
- 注(2)ろ過は分離型ろ過器を用いて行い、ろ紙はあらかじめ水でよく洗浄しておく。
- (3)湿泥で2~5gを目途に採取する。
- (4)ろ過により空気にさらされるので、手早く操作して硫化物の消失を防ぐ。
- (5)試料が砂質の時は突沸することがあるので、フラスコ容量の大きいものを用いる。
- (6)留出速度2.5~3ml/minで蒸留を行う。留出速度が速すぎると硫化水素が完全に吸収され在い。特に蒸留開始時は留出速度を遅くして損失を防ぐ。
- (7)ガラス管は1回ごとに交換する。留出過程でガラス管に付着した硫化物は、滴定操作において塩酸酸性にすれば溶解するので、一緒に滴定する。
(5)操作
- (a)(4)で調製した試験溶液によう素溶液(N/100)25ml、塩酸(1+1)2mlを加えてよく振り混ぜる(注8)。
- (b)残ったよう素をN/100チオ硫酸ナトリウム溶液で滴定し、よう素の黄色が薄くなったら、指示薬としてでんぶん溶液約1mlを加え、よう素でんぶんの青色が消えたときを終点とする(注9)。
- (c)空試験は(4)で調製した空試験溶液について(a)~(b)の操作を行う(注10)。
- (d)次式によって乾燥試料1g当りの硫化物態硫黄量(mgS/g)を算出する。

- 注(8)よう素溶液を加えてから塩酸を加える。逆に行うと硫化水素として損失するおそれがある。
- (9)硫化物が多量に含まれる場合は、よう素溶液(N/10)及びチオ硫酸ナトリウム溶液(N/10)を用いる。
- (10)空試験は蒸留操作を省き、滴定のみでもよい。
18 全窒素(ケルダール窒素)
18.1中和滴定法
(1)試薬
- (a)N/20硫酸 硫酸1.5mlをあらかじめ水100mlを入れたビーカーに加えてよくかさ混ぜ、水で1㍑とする。
標定 炭酸ナトリウム(無水)(標準試薬)を500~650℃で40~60分間加熱し、デシケー夕ー中で放冷した後、その0.5300gをはかり取り、水に溶かして全量フラスコ200mlに入れ、水を標線まで加える。この溶液20mlをビーカー300mlにとり、指示薬としてメチルレッド-プロムクレゾールグリーン混合溶液3~5滴を加え、この硫酸(N/20)で滴定する。終点付近に達したら煮沸して二酸化炭素を追い出し、冷却後、溶液の色が灰紫を呈するまで滴定する。次式によってN/20硫酸のファクター(f)を算出する。

- (b)硫酸カリウム
- (c)硫酸鋼五水和物 粉末
- (d)ほう酸溶液(飽和)ほう酸50gに水1㍑を加えて振り混ぜ、その上澄み液を用いる。
- (e)メチルレットプロムクレゾールグリーン混合溶液 メチルレッド0.02gとプロムクレゾールグリーン0.10gとをエタノール(95)100mlに溶かす。
- (f)水酸化ナトリウム溶液(50w/v%)水酸化ナトリウム250gを水に溶かして500加とする。使用時に調製する。
(2)装置
(a)蒸留装置 図18.1に一例を示す。
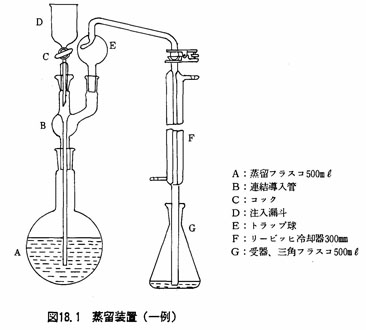
(3)試験溶液の調製
- (a)2.1湿試料の適量(注1)を0.01gのけたまではかり取り、少量の水でケルダールフラスコ200mlに移す。
- (b)これに硫酸10ml、硫酸カリウム5g及び硫酸鋼五水和物2gを加え、加熱して硫酸の白煙を発生させ、引き続き30分間強熟して有機物を分解する。
- (c)放冷後、少量の水を加えてよく振り混ぜる。不溶解物が沈降するのを待って傾斜法により上澄み液を全量フラスコ200mlに移す。ケルダールフラスコの内壁及び不溶解物を水で洗浄し、再び傾斜法により上澄み液を全量フラスコ中に合わせる。この操作を繰り返した後、水を標線まで加える。
- (d)この中から適量を蒸留フラスコに移し、沸騰石数個及び水を加えて液量を約300mlとする。
- (e)蒸留フラスコを図18.1のように連結し、受器には三角フラスコ500mlを用い、これにほう酸飽和溶液50lmを加え、指示薬としてメチルレッドープロムクレゾールグリーン混合溶液5~7滴を加えておく。
- (f)蒸留フラスコ上部の漏斗から水酸化ナトリウム(50w/v%)を適量(注2)加えた後、蒸留フラスコを加熱し、留出速度5~7ml/minで蒸留を行う(注3)。
- (g)約140mlが留出したら蒸留を止める。
- (h)冷却器と逆流止めを取り外し、冷却器の内管及び逆流止めの内外を少量の水で洗う。洗液は受器の三角フラスコ500mlに合わせる。これを試験溶液とする。
- (i)別に水30mlをとり、(b)~(h)の操作を行い空試験溶液とする。
- 注(1)窒素として0.23~30mgを含むようにとる。風乾により窒素の値が変化する恐れのない試料では2.2風乾試料を用いてもよい。
- (2)(d)での分取量100mlあたり20mlの割合で加える。
- (3)冷却器の管の先端は、常に液面下15mm以上に保つようにする。
備考 1 蒸留法として水蒸気蒸留法を用いてもよい。その場合は、図18.1の蒸留フラスコに水蒸気を送るように装置を組み立て、蒸留フラスコを加熱し、沸騰し始めたら水蒸気を蒸留フラスコに送り、留出速度3~5ml/minで蒸留し、約140mlが留出したら水蒸気を止める。
(4)操作
- (a)試験溶液の全量を用い、N/20硫酸溶液で溶液の色が灰紫を呈するまで滴定する。
- (b)別に空試験として、(3)(i)の溶液について(a)の操作を行い、試験溶液について得た滴定値を補正する。
- (c)次式によって試料中の窒素の濃度(mgN/g)を算出する。

備考 2 中和滴定法において試験溶液中のアンモニウムイオンの濃度が低い場合は、N/20硫酸の代わりにN/50硫酸溶液(注4)を用いてもよい。この場合、次式によって試料中の窒素の濃度を算出する。

- 注(4)(1×a)N/20硫酸100mlを全量フラスコ250mlに取り、水を標線まで加える。この溶液は使用時に調製する。
- (5)(1)(a)N/20硫酸溶液のファクターを用いる。
備考 3 (3)(e)のほう酸飽和溶液の代わりに硫酸(N/20)を用いてもよい。この場合は次のように操作する。 L
三角フラスコ500mlに硫酸(N/20)(注8)50mlを正しく加え、指示薬としてメチルレッドープロムクレゾールグリーン混合溶液5~7滴を加え、(3)(e)~(h)の操作を行う。
次に、N/20水酸化ナトリウム溶液(注7)で溶液の色が灰紫を呈するまで滴定する。別に空試験として、(3)(i)の溶液についてこの操作を行い、試験溶液について得た滴定値を補正する。更にこれとは別に硫酸(N/20)50mlを正しく三角フラスコ500mlにとり、水150mlを加え、指示薬としてメチルレッドープロムクレゾールグリーン混合溶液5~7滴を加え、以下試験溶液の場合と同様に滴定を行い硫酸(N/20)50mlに相当するN/20水酸化ナトリウム溶液のml数を求める。
次式によって試料中の窒素の濃度(mgN/g)を算出する。

- 注(6)18.1(1)(a)N/20硫酸の標定は行わずに用いる。
- (7)N/20水酸化ナトリウム溶液 水約30mlをポリエチレンびんにとり、水酸化ナトリウム約35gを加えて溶かし、密栓して一夜放置する。その上澄み液2.5mlをとり、全量フラスコ1000mlに入れ、炭酸を含まない水を標線まで加える。
標定 あらかしめデシケーター中に約48時間放置して乾燥したアミド硫酸(スルファミン酸)(標準試薬)約1gを0.1mgまではかり取り、水に溶かして全量フラスコ200mlに入れ、水を標線まで加える。その20mlを三角フラスコ300mlにとり、指示薬としてプロムチモールブルー溶液(0.1w/v%)〔プロムチモールブルー0.1gをエタノール(95)に溶かし、水で100mlとする。〕3~5滴を加え、このN/20水酸化ナトリウム溶液で滴定し、溶液の色が緑になったときを終点とする。次式によってN/20水酸化ナトリウム溶液のファクター(f)を算出する。

18.2 インドフェノール青吸光光度法
(1)試薬
- (a)硫酸カリウム
- (b)硫酸銅五水和物18.1(1)(c)と同じ。
- (c)ほう酸溶液(飽和)18.1(1)(d)と同じ。
- (d)水酸化ナトリウム溶液(50w/v%)18.1(1)(f)と同じ。
- (e)水酸化ナトリウム溶液(20w/v%) 水酸化ナトリウム20gを水に溶かして100mlとする。この溶液は使用時に調製する。
- (f)ナトリウムフェノラート溶液 フェノール25gを水酸化ナトリウム(20w/v%)55mlに溶かし、放冷後、アセトン6mlを加え、水で200mlとする。使用時に調製する。
- (g)次亜塩素酸ナトリウム溶液(有効塩素1w/v%) 次亜塩素酸ナトリウム溶液(有効塩素5~12%)の有効塩素の濃度を定量し(注8)、有効塩素量が約1w/v%になるように水で薄める。使用時に調製する。
- (h)窒素標準液(1mgN/ml)過塩素酸マグネシウムを入れたデシケーター中で一夜放置した塩化アンモニウム3.82gを水に溶かして全量フラスコ1000mlに入れ水を標線まで加える。0~10℃の暗所に保存する。
- (i)窒素標準液(0.01mgN/ml) 窒素標準液(1mgN/ml)10mlを全量フラスコ1000mlにとり、水を標線まで加える。
注(8)次亜塩素酸ナトリウム溶液10mlを全量フラスコ200mlにとり、水を標線まで加える。この10mlを共栓三角フラスコ300mlにとり、水を加えて約100mlとする。よう化カリウム1~2g及び酢酸(1+1)6mlを加えて密栓し、よく振り混ぜて暗所に約5分間放置した後、N/20チオ硫酸ナトリウム溶液で滴定する。溶液の黄色が薄くなったら、指示薬としてでんぶん溶液約2mlを加え、生じたよう素でんぷんの青い色が消えるまで滴定する。別に空試験として10mlをとり、同じ操作を行って滴定値を補正する。

(2)装置
18.1(2)と同じ。
(3)試験溶液の調製
- (a)18.1(3)(a)~(d)の操作を行う(注9)。
- (b)蒸留フラスコを図18.1のように連結し、受器には共栓メスシリンダー200mlを用いほう酸(飽和)溶液50ml(注 10)を入れる。
- (c)18.1(3)(f)~(h)の操作を行う。ただし、洗液は受器の共栓メスシリンダー200mlに合わせ、水を200mlの標線まで加える。
- (d)別に水30mlをとり、18.1(3)(b)~(h)の操作を行い、空試験溶液とする。
- 注(9)窒素として0.032mg以上を含むように試料をとる。
- (10)ほう酸溶液の代わりに硫酸(N/20)50mlを用いてもよい。
(4)操作
- (a)試験溶液の適量(窒素として0.004~0.08mgを含む。)を全量フラスコ50mlにとり、水を加えて約25mlとする。
- (b)ナトリウムフェノラート溶液10mlを加えて振り混ぜる。
- (c)次亜塩素ナトリウム溶液(有効塩素1w/v%)5mlを加え、水を標線まで加え、栓をして振り混ぜる。
- (d)液温を20~25℃に保って約30分間放置する(注11)。
- (e)この溶液の一部を吸収セルに移し、波長630nm付近の吸光度を測定する。
- (f)(3)(d)空試験溶液について(a)~(e)の操作を行って吸光度を求め、試験溶液について得た吸光度を補正する。
- (g)検量線から窒素の量を求め、分析試料中の窒素の濃度を算出する。
- (h)3乾燥減量で求めた分析試料の乾燥減量(%)で(g)の窒素濃度を補正し、乾燥試料当りの濃度(mgN/g)として結果を表示する。
検量線 窒素標準液を段階的に(窒素として0.004~0.08mg)全量フラスコ50mlに取り、水を加えて約25mlとし、(b)~(e)の操作を行って吸光度を測定し、窒素の量と吸光度との関係線を作成する。
注(11)液温が20~25℃のとき約30分間で発色は最高となり、その後30分間は安定である。
19 全りん
19.1硝酸一過塩素酸分解法
(1)試薬
- (a)モリブデン酸アンモニウム溶液セモリプデン酸六アンモニウム四水和物6gとタルトラトアンチモン(Ⅲ)酸カリウム0.24gを水約300mlに溶かし、これに硫酸(2+1)120mlを加え、次にアミド硫酸アンモニウム5gを溶かした後、水を加えて500mlとする。
- (b)L-アスコルビン酸溶液(7.2w/v%)L-アスコルビン酸7.2gを水に溶かして100mlとする。0~10℃の暗所に保存する。色の着いた溶液は使用しない。
- (c)モリブデン酸アンモニウム-アスコルビン酸混合溶液 モリブデン酸アンモニウム溶液及びL-アスコルビン酸溶液を5対1の体積比で混合する。使用時に調製する。
- (d)りん標準液(0.05mg/ml)りん酸二水素カリウムを110℃で約3時間乾操し、デシケーター中で放冷した後、その0.2197gをとり適量の水に溶かして全量フラスコ1000mlに入れ、水を標線まで加える。0~10℃の暗所に保存する。
- (e)りん標準液(0.005mg/ml)りん標準液(0・05mg/ml)25mlを全量フラスコ250mlにとり、水を標線まで加える。使用時に調製する。
(2)試験溶液の調製
- (a)2.1湿試料の適量(注1)を0.01gのけたまではかり取り、少量の水でビーカーに移す。
- (b)硝酸5mlを加えて熱板上で静かに加熱して15~20mlに濃縮する。
- (c)これに硝酸5mlを加えて再び加熱し、約10mlになるまで濃縮する。
更に硝酸5mlを加えて加熱し、約10mlになるまで濃縮した後、放冷する。 - (d)過塩素酸(60%)5mlを少量ずつ加える。熱板上で加熱を続け、過塩素酸の白煙が発生し始めたらビーカーを時計皿で覆い、過塩素酸の白煙がビーカーの内壁を還流する状態に保つ(注2)。
- (e)放冷後、ビーカーの内壁を少量の水で洗浄し、水約30mlを加えて静かに加熱する。
- (f)不溶解物が沈降するのを待って、ろ紙5種Bでろ過する。ビーカーの中の不溶解物及びろ紙を温水で洗浄する。ろ液と洗液を合わせて室温まで冷却し、全量フラスコ100mlに移し入れ、水を標線まで加え、これを試験溶液とする。
- 注(1)2.2風乾試料又は2.3乾燥試料を用いてもよい。試料量は乾操試料として約1gを目途にはかり取るとよい。
- (2)この操作によっても有機物が分解されず、溶液に色が残った場合には、硝酸5mlを加えて加熱する操作を繰り返す。
(3)操作
- (a)試験溶液の適量(りんとして0.005~0.04mlを含む)を全量フラスコ50mlにとる。
- (b)この溶液に、まず水酸化ナトリウム溶液(20w/v%)を、次に水酸化ナトリウム(4w/v%)を加える。水酸化ナトリウム(4w/v%)の添加は、溶液に金属水酸化物の沈殿が生じる直前にとどめる。必要に応じて硫酸(1+35)を用いて調節する(注3)。
- (c)水を加えて約40mlとした後、モリプデン酸アンモニウム-アスコルビン酸混合溶液3.5mlを加えて振り混ぜる。水を標線まで加え、20~40℃で15分間放置する(注4)。
- (d)溶液の一部を吸収セルに移し、波長880nm又は710nmの吸光度を測定する。
- (e)全操作にわたって空試験を行い、試験溶液について得た吸光度を補正する。
- (f)検量線からりんの量を求め、分析試料中のりんの濃度を算出する。
- (g)3乾燥減量で求めた分析試料の乾燥減量(%)で(f)のりん濃度を補正し、乾燥試料当りの濃度(mgP/g)として結果を表示する。
検量線 りん標準液を段階的に(りんとして0.005~0.04mgを含む)とり、(a)~(d)の繰作を行って吸光度を測定し、りんの量と吸光度との関係線を作成する。
- 注(3)金属水酸化物の沈殿が少なくpH調整が困難な時は、P-ニトロフェノール溶液(0.1w/v%)〔p-ニトロフェノール0.1gを水に溶かして100mlとする〕数滴を加え、溶液がわずかに黄色を示すまで中和する。
- (4)検量線作成時と同じ発色温度となるようにする。
備考 1試料中にひ素(Ⅴ)が含まれると、りん酸イオンと同様に発色して妨害し、ひ素1μgはりん約0.35μgに相当する。この時は、別に13ひ素により定量し、補正する。
2 鉄(Ⅱ)30mg以上は、モリプデン青を退色させる。L-アスコルビン酸溶液の添加量を増せば妨害を抑制できる。
19.2硝酸-硫酸分解法
(1)試薬
19.1(1)と同じ。
(2)試験溶液の調製
- (a)19.2(2)(a)及び(b)の操作を行う。
- (b)(a)の操作後の溶液に硫酸5ml及び硝酸5mlを加え、加熱して硝酸の白煙が発生するまで濃縮し、更に加熱して硫酸の白煙を短時間強く発生させた後、放冷する。
- (c)この溶液に硝酸5mlを加え、再び硫酸の白煙が発生するまで加熱する (注2)。
- (d)放冷後水約30mlを加え、約10分間静かに煮沸する。
- (e)以下19.1(2)の(f)の操作を行う。これを試験溶液とする。
(3)操作
19.1(3)と同じ。
20 過マンガン酸カリウムによる酸素消費量〔CODsed(底質(Sediment)のCODのこと。)〕
(1)試薬
- (a)過マンガン酸カリウム溶液(N/10)過マンガン酸カリウム3.2gを平底フラスコにとり、水1050~1100mlを加えて溶かす。これを1~2時間静かに煮沸した後、一夜放置する。
上澄み液をガラスろ過器G4を用いてろ過する(ろ過前後に水洗しない)。ろ液は約30分間水蒸気洗浄した着色びんに入れて保存する。 - (b)しゅう酸ナトリウム溶液(N/10)しゅう酸ナトリウム6.8gを水に溶かして1㍑とする。
- (c)でんぶん溶液 でんぶん(溶性)1gを水約10mlに混ぜ、熱水100ml中にかき混ぜながら加え、約1分間煮沸した後、放冷する。使用時に調製する。
- (d)よう化カリウム溶液(10w/v%)よう化カリウム10gを水に溶かして100mlとする。着色びんに入れ時所に保存する。溶液に色の付いたものを用いてはならない。
- (e)N/10チオ硫酸ナトリウム溶液 チオ硫酸ナトリウム五水和物26g及び炭酸ナトリウム(無水)0.2gをとり、水に溶かして1㍑とし、これに3-メチル-1-ブタノール(イソアミルアルコール)約10mlを加え、よく振り混ぜた後2日間放置する。
標定 よう素酸カリウム(標準試薬)を120~140℃で2時間乾燥し、デシケ-ター中で放冷した後0.7134gをとり、水に溶かし、全量フラスコ200mlに入れ、水を標線まで加える。この20mlをとり、共栓三角フラスコ300mlに入れ、よう化カリウム2g及び硫酸(1+5)5mlを加え、直ちに栓をして静かに振り混ぜ、暗所に5分間放置する。
水100mlを加え、遊離したよう素をこのチオ硫酸ナトリウム溶液で滴定する。溶液の黄色が薄くなってから、指示薬としてでんぶん溶液1mlを加え、よう煮でんぶんの青い色が消えるまで滴定する。
別に、同一条件で空試験を行って補正したml数から次式によってN/10チオ硫酸ナトリウム溶液のファクター(f)を算出する。

(2)操作
- (a)2.1湿試料(注1)の適量(注2)を三角フラスコ300ml(注3)に0.01gのけたまではかり取り、過マンガン酸カリウム溶液(N/10)を正確に100ml、水酸化ナトリウム溶液(30w/v%)5mlを加え、よく振り混ぜる。
- (b)沸騰水溶中に入れ、30分間加熱する(注4)。
- (c)加熱終了後、直ちにしゅう酸ナトリウム溶液(N/10)(注5)を正確に100ml、硫酸(3+7)10mlを加えて過マンガン酸カリウムの色を褐色させ、室温まで冷却する。
- (d)三角フラスコの内容物を共栓メスシリンダー500mlに水で洗い流し、水を標線まで加え、よく振り混ぜる。
- (e)乾燥ろ紙を用いてろ過し、ろ液100mlを三角フラスコ300mlにとり、過マンガン酸カリウム溶液(N/10)を正確に10ml加え、かき混ぜながら数分間放置する(注6)。
- (f)よう化カリウム溶液(10w/v%)5mlを加えてよく振り混ぜる。
- (g)遊離したよう素をN/10チオ硫酸ナトリウム溶液で滴定し、溶液の色が薄い黄色になったら、指示薬としてでんぶん溶液1mlを加え、よう素でんぷんの青い色が消えるまで滴定を続ける。
- (h)空試験は試薬だけを用いて、(a)~(g)の操作を行う。
- (i)別に、3乾燥減量によって乾燥減量(%)を求め、次式によって乾燥試料1g当りのCODsed(mgO/g)を算出する。

- 注(1)乾燥によりCODsed値が変化する恐れのない試料では、2.2風乾試料を用いてもよい。
- (2)試料の採取量によって分析値が大きく変動するため、試料は最初に加えた過マンガン酸カリウムの40~60%が加熱中に消費されるように採取する。このため、あらかじめ試料を段階的にとり、予備試験を実施する。被酸化性物質の量が少ない場合の採取量は最大10gでよい。
- (3)三角フラスコの容量、形状により分析値が変化するので、注意する。
- (4)試料加熱時の水浴は常に沸騰状態を維持し、試料の液面は沸騰水浴の水面下で、かつ、三角フラスコが水溶の底に直接接しないように保つ。
- (5)過マンガン酸カリウム溶液(N/10)よりしゅう酸ナトリウム溶液(N/10)の濃度の方をやや濃くしておく。
- (6)過マンガン酸カリウム溶液(N/10)10mlを加えても全部消費されて、過マンガン酸カリウムの色が消失した場合には、更に10mlを追加する。この場合、空試験においても過マンガン酸カリウム溶液(N/10)20mlを用いる。
備考 1(c)以下の操作を以下のように行ってもよい。ただし、滴定の終点が不鮮明な場合は、この方法は用いない。
操作
(b)で得られた加熱終了後の溶液に、直ちによう化カリウム溶液(10w/v%)25mlを加えて振り混ぜた後、室温まで冷却する。
次に、硫酸(3+7)10mlを加えて振り混ぜた後、遊離したよう素をN/4チオ硫酸ナトリウム溶液で滴定する。滴定操作は、溶液の茶褐色が薄くなったら指示薬としてでんぶん溶液を2ml加え、よう素でんぶんの青色が消え、灰白色となった点を終点とする。
空試験は試薬だけを用いて同様に操作する。
次式によって、CODsed(mgO/g)を算出する。

2 CODsed試験は反応条件により分析値が変動するので注(2)~(4)を守らなければならない。その場合でも、一定した分析値が得にくい試料もある。
Ⅲ 溶出試験
1 溶出率の算定法
溶出率は次式で求めるものとする。

W1:溶出試験に使用した分析試料中の有害物質量(μg)
W2:溶出試験に使用した混合液の体積に相当する溶出液中に含まれる有害物質量(μg)
2 水銀
2.1総水銀(注1)
注(1)この試験は乾燥固形分当たり総水銀含有量10ppm(mg/kg)以上のものについて適用する。
(1)試薬
昭和46年12月環境庁告示第59号(注2)付表3の1試薬に準ずる。注(2)以下「告示」という。
(2)器具及び装置
告付表3の2器具及び装置に準ずる。
(3)試験溶液の調製
- (a)混合液中に含まれる乾燥固形分の質量と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるように、Ⅱ2・1の湿試料を取り(注3)、水を加えて混合液を調製する。
- (b)室温において4時間連続して、かき混ぜ又は振り混ぜる。
- (c)約30分間放置したのち、ろ紙5種Cを用いてろ過し、ろ液(浮出液)を試験溶液とする。
注(3)試料は、混合液の体積の約2倍の容量の容器にとる。容器は、水銀の溶出や吸着のない材質で、密栓ができる形のものを用いる。
(4)操作
- (a)試験溶液について、告示付表三の4試験操作及び5検量線の作成に準じて行い、溶出液に含まれる水銀量を求める。
- (b)(3)(a)で使用した試料(湿試料)中の水銀量(4)と(a)で求めた溶出液中に含まれる水銀量とから、1溶出率の算定法にしたがって水銀の溶出率を計算する。
注(4)分析試料の水銀濃度が乾燥試料当りの濃度で示されている場合は、次式にしたがって湿試料当りの濃度に換算したのち、使用した分析試料(湿試料)中の水銀量を算出する。
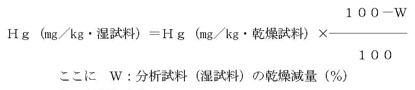
2.2 アルキル水銀化合物
(1)試薬
告示付表4 第1 試薬に準ずる。
(2)器具及び装置
告示付表4 第1 器具及び装置に準ずる。
(3)試験溶液の調製
- (a)混合溶液中に含まれる乾燥固形分の重量と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるようにⅡ2・1の湿試料を取り、水を加えて混合液を調製する。
- (b)室温において4時間連続して、かさ混ぜ又は振り混ぜる。
- (c)約30分放置した後、ろ紙5種Cを用いてろ過し、ろ液(浮出液)を試験溶液とする。
(4)操作
- (a)試験溶液について、告示付表4第14試験操作及び5 検量線の作成に準じて行い、溶出液に含まれるアルキル水銀量を求める。
- (b)(3)(a)で使用した試料(湿試料)中のアルキル水銀量(注1)と(a)で求めた溶出液中に含まれるアルキル水銀量とから1溶出率の算定法にしたがってアルキル水銀化合物の溶出率を計算する。
注(1)2.1給水銀量の(4)の注(4)と同様に湿試料中のアルキル水銀量を算出する。
3 カドミウム
(1)試薬
JIS K0102(工場排水試験方法)(注1)55.2(1)試薬に準ずる。
注(1)以下規格という。
(2)器具及び装置
規格55.2(2)器具及び装置に準ずる。
(3)試験溶液の調製
- (a)混合液中に含まれる乾燥固形分の質量と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるようにⅡ2.1の湿試料を取り、水を加えて混合液を調製する。
- (b)室温において、4時間連続して、かき混ぜ又は振り混ぜる。
- (c)約30分間放置した後、ろ紙5種Cを用いてろ過し、ろ液(溶出液)を試験溶液とする。
(4)操作
- (a)試験溶液について、規格55.2(4)操作に準じて行い、溶出液中に含まれるカドミウム量を求める。
- (b)(3)(a)で使用した試料(湿試料)中のカドミウム量(注2)と(a)で求めた溶出液中に含まれるカドミウム量とから1溶出率め算定法にしたがってカドミウムの溶出率を計算する。
注(2)2.1総水銀(4)の注(4)と同様に湿試料中のカドミウム量を算出する。
4 鉛
(1)試薬
規格54.2(1)試薬に準ずる。
(2)器具及び装置
規格54.2(2)器具及び装置に準ずる。
(3)試験溶液の調製
3カドミウム(3)に準ずる。
(4)操作
- (a)試験溶液について、規格54.2(4)操作に準じて行い、溶出液中に含まれる鉛量を求める。
- (b)(3)(a)で用いた試料(湿試料)中の鉛量(1)と(a)で求めた溶出液中に含まれる鉛量とから1溶出率の算定法にしたがって鉛の溶出率を計算する。
注(1)2.1総水銀(4)の注(4)と同様に湿試料中の鉛量を算出する。
5 ひ素
(1)試薬
- (a)過マンガン酸カリウム溶液(0.3w/v%)過マンガン酸カリウム0.3gを水に溶かして100mlとする。
- (b)メタクレゾールバープル溶液(0.1w/v%)メタクレゾールバーブル溶液0.1をエタノール.(95)50mlに溶かし、水を加えて100mlとする。
- (c)鉄(Ⅱ)溶液 塩化鉄(Ⅱ)六水和物5g又は硫酸鉄(Ⅱ)アンモニウム十二水和物9gを塩酸5mlと水に溶かして100mlとする。
- (d)測定用試薬 規格61.1(1)又は61.2(1)と同じ。
(2)試験溶液の調製及び濃縮
- (a)3カドミウム(3)に準ずる。
- (b)試験溶液の適量をとり、試験溶液1ゼにつき硝酸3mβと過マンガン酸カリウム溶液(0.3w/v%)とを滴加して着色させる。
- (c)これを煮沸して過マンガン酸が分解して脱色されたときは、再び過マンガン酸カリウム溶液(0.3w/v%)を脱色されなくなるまで加える。
- (d)過酸化水素水(1+30)のできるだけ少量を滴加して過剰の過マンガン酸を分解する。
- (c)鉄(Ⅱ)溶液1ml、メタクレゾールバーブル溶液(0.1w/v%)2~3滴を加え、液温約80℃でかき混ぜながらアンモニア水(1+21)を溶液の色が紫(pH約9)になるまで滴加する。
- (f)放置して沈殿が沈降した後、小さいろ紙でろ別し、温水で2~3回洗浄する。
(3)操作
- (a)(2)(f)で得た沈殿を仕るべく少量の水で水素化ひ素発生びんに洗い入れ温硫酸(1+5)18mlと塩酸(1+1)2mlに溶かして、水で約40mlとする。よう化カリウム溶液(20w/v%)15ml及び塩化すず(Ⅱ)溶液5mlを加えて振り混ぜ10分間放置した後、規格61.1の(3)操作の(e)から(i)の操作を行って溶出液中に含まれるひ素量を求める。
又は、(2)(f)で得た沈殿を少量の水で反応容器に洗い入れ、温塩酸(1+4)10mlに溶かして、水で約20mlとし(注1)、規格61.2の(3)操作の(d)(注2)から(h)の操作を行って溶出液中に含まれるひ素量を求める。 - (b)(2)で用いた分析試料(湿試料)中のひ素量(3)と(a)で求めた溶出液中に含まれるひ素量とから1溶出率の算定法にしたがってひ素の溶出率を計算する。
- 注(1)沈殿をビーカーに洗い入れて溶解し、全量フラスコで定容としてから分取してもよい。この場合は、改めて塩酸濃度を調製する。
- (2)鉄(Ⅱ)溶液は加えない。
- (3)2.1総水銀(4)の注く4)と同様に湿試料中のひ素量を算出する。
6 ヘキサクロロシクロヘキサン(HCH、旧称BHC)
(1)試薬
Ⅱ分析方法16ヘキサクロロシクロヘキサン(HCH)の(1)試薬と同じ。
(2)試験溶液の調製
- (a)混合液中に含まれる乾燥固形分の質量と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるように分析試料(湿試料)を取り、水を加えて混合液を調製し、室温において4時間連続して、かき混ぜ又は振り混ぜる。
- (b)約30分放置したのち、ガラス繊維ろ紙を用いてろ過する。ろ液から100mlを分液漏斗にとり、飽和食塩水6mlとヘキサン50mlを加え、激しく5分間振り混ぜる。
- (c)ヘキサン層を分離し、水層に再びヘキサン50mlを加えて激しく振り混ぜる。ヘキサン層を分離し、先のヘキサンと合わせる。
- (d)ヘキサン層を分液晶斗に入れ水100mlで2回洗浄する。
- (e)以下、Ⅱ分析方法16(3).1(d)以下の操作に準じて試験溶液を調製する。
(3)操作
(a)Ⅱ分析方法16(4).1ガスクロマトグラフによるHCHの測定法に準して操作し、溶出液中に含まれるHCH4異性体の合量を次式により求める。

(2)(a)で使用した分析試料(湿試料)中のHCH量(注1)(注2)と(a)で求めた溶出液中に含まれるHCH量とから1溶出率の算定法にしたがってHCHの溶出率を計算する。
- 注(1)2.1総水銀の(4)の注(5)と同様にする。
- (2)HCH量は4異性体(α、β、γ、σ)の合量を求める。