

法令・告示・通達
底質調査方法について
環水管120号
(環境庁水質保全局長から各都道府県知事・権限委任市長あて通達)
各種の公共用水域等の底質調査について、別添「底質調査方法」のとおり分析方法等を定めたので通知する。
なお、別添「底質調査方法」は、通常の底質調査における分析方法等を定めたものであり、特殊な条件のもとで、これによることが著しく不適当と認められる場合は、これの骨子に沿つて必要な変更を行つても差し支えない。
おつて、関係者に対して、この趣旨の周知徹底方を図られたい。
別表
底質調査方法
1 採泥
1) 採泥時期
底質中に含まれる物質が、水利用に悪影響を及ぼす時期を含めることを原則とし、当該水域につき水質調査の実施が予定されている場合は、水質調査の実施時期に合わせることが望ましい。
2) 採泥地点
(1) 概況調査
海域、湖沼においては、調査対象水域の規模及び予想される汚染の程度に応じて均等に2~6kmメツシユで採泥地点を設けるものとする。主要な排水口周辺水域等においては、地点を増加する。
河川においては、原則として、主要な排水口の直下50m下流及び流下方向1kmごとの汚泥の堆積しやすい地点とする。水域の状況等により適宜地点を増加する。
(2) 精密調査
海域、湖沼においては調査対象水域に200~300mメツシユで採泥地点を設定するものとし、河口部等の堆積汚泥の分布状況が変化しやすい場所等においては必要に応じて地点を増加するものとする。
河川及び水路においては、幅の広いときにあつては50mメツシユで、幅の狭いときにあつては流下方向50mごとに汚泥の堆積しやすい場所を採泥地点とし、水域の状況等により適宜地点を増加する。
3) 採泥方法
2)の各採泥地点において、エクマンバージ型採泥器又はこれに準ずる採泥器によつて3回以上底質を採取し、それらを混合して採泥試料とする。
ただし、概況調査の場合は、SK式採泥器又はこれに準ずるものを用いても差し支えない。深さ方向の調査が必要な場合には、柱状試料を採取することとし、この場合は、原則として底質表面から深さ1mごとの各位置において、その各10cm程度の泥を採取し、その位置の試料とする。なお採取は1回でも差し支えない。
SK式採泥器を用いる場合には、なるべく短時間で1回採取を行いそのものを試料とする。
4) 採泥時に実施すべき事項
採泥日時、採泥地点(図示すること)、採泥方法(使用した採泥器の型名)、底質の状態(堆積物、砂、泥などの別、色、臭気など)及びpHは直ちに観測測定し記録する。試料はできるだけ速やかに分析する。直ちに分析が行えない場合には、温度を低く保つておくこととする。なお、調査の目的に応じてその他の項目を適宜追加する。
5) 採泥時の試料の調製
採泥試料を清浄なポリエチレン製のパツト等(測定重金属等の物質の吸着、溶出等がない材質のものを使用する。)に移し、小石、貝殼、動植物片などの異物を除いた後、均等に混合し、その500~1,000gを清浄なポリエチレンびん、ポリエチレン袋等(測定重金属等の物質の吸着、溶出等がない材質のものを使用する。)に入れて、実験室に持ち帰るものとする。
2 分析方法
1) 分析時の試料の調製
1―5)によつて持ち帰つた試料から、適量の代表試料を分取し、これを3,000rpmで20分間遠心分離し、その沈澱固形物を分析試料とする。
2) 分析値の表示
底質試料中の重金属等有害物質は、乾燥固形物当りの濃度に換算し、有効数字3桁まで表示するものとする。
3) 乾燥方法
2―1)の分析試料から5g以上(a)を重量既知の共せん付はかりびんに正確にはかり取り、105~110℃で恒量になるまで乾燥し(通常約2時間)デシケーター中で放冷し、固形物の重量(b)を求め、次式によつて水分含有率(%)を算出する。
水分含有率(%)=((a-b)/a)×100
a:始めに採取した分析試料の重さ(g)
b:乾燥後の固形物の重さ(g)
4) 強熱減量
2―3)で乾燥した泥を正確に5g以上を大型のるつぼにはかり取り、電気炉を用いて600±25℃で恒量になるまで強熱し(通常約2時間)デシケーター中で放冷し、固形物の重量を求める。
始めに測定した重量から終わりの重量を差し引き採取した試料に対する重量百分率を計算する。
5) 総水銀
(1) 試薬
塩化第一すず溶液:塩化第一すず(SnCl2・2H2O)10gに硫酸(H2SO4)(1+20)60mlを加え、かきまぜながら加熱して溶かし、冷却後水で100mlとする。
ジチゾン―四塩化炭素溶液(0.01W/V%):精製した新しいジチゾン(ジフエニルチオカルバゾンC6H5NNCSNHNHC6H5)0.11gを四塩化炭素(CCl4)400mlによくかき混ぜて溶かし、ろ紙でろ過する。この溶液を分液ロートに移し入れ、アンモニア水(NH4OH)(1+100)400mlを加え、振り混ぜてジチゾンを水層に移し、静置した後四塩化炭素層を分離する。水層に四塩化炭素50mlを加えて振り混ぜ、洗浄する。静置後、四塩化炭素層を分離し、この洗浄を四塩化炭素層が薄い緑となるまで繰り返す。洗浄した水層に四塩化炭素500mlと塩酸(HCl)(1+10)50mlとを加えて振り混ぜ、ジチゾンを四塩化炭素層に移す。
静置後、四塩化炭素層を分離する。水層には四塩化炭素50mlを加えて振り混ぜ、残つたジチゾンを抽出する。静置後先のジチゾン―四塩化炭素溶液に加え、更に四塩化炭素を加えて1lに薄め、着色びんに入れ、飽和亜硫酸水100mlを加えて表面をおおい10℃以下で暗所に保存する。
ジチゾン―四塩化炭素溶液(0.005W/V%):ジチゾン四―塩化炭素溶液(0.01W/V%)を四塩化炭素で2倍に薄める。
水銀標準原液:塩化第二水銀(HgCl2)0.677gを水に溶かし、メスフラスコ1lに入れ、水を標線まで加える。この溶液1mlは水銀0.5mgを含む。
水銀標準溶液:水銀標準原液を水を用いて適当な濃度となるように調製する。
(2) 試験溶液の調製
2―1)の分析試料約10g(湿泥)を正確にはかりとり、これを還流冷却器付フラスコに入れ、硝酸(1+1)50mlで有機物を分解する。その後室温まで冷却して過マンガン酸カリウム溶液(6W/V%)10mlを加えて1時間加熱還流する。もしこの間に過マンガン酸カリウムの色が消失する場合は、室温まで冷却した後過マンガン酸カリウム溶液(6W/V%)5mlを追加して、再び加熱する。この操作を赤紫色が約10分間残るまで繰返す。液温を約40℃に冷却し、尿素溶液(10W/V%)10mlを加え溶液を振り混ぜながら、塩酸ヒドロキシルアミン溶液(20W/V%)を滴加し、過剰の過マンガン酸カリウムを分解する。これをガラス綿又はガラス繊維ろ紙でろ過し、ろ液が微紅色になるまで過マンガン酸カリウム溶液(6W/V%)を滴加し、水を加えて定容とし、これを試験溶液とする。
(3) 測定方法(原子吸光光度法)
a) 還元気化循環法
試験溶液の適量(総水銀として0.1~1.0μgに相当する量)を取り、これに硫酸(1+1)10mlを加え、原子吸光装置付属の検水びんに移し入れ、液量を水を加えて100mlとする。次に、塩化第一すず溶液10mlを加え、直ちに装置に連結し、循環ポンプを作動させ、空気を循環させる。記録計の指示が急速に上昇して一定値を示すまでポンプの作動を続ける。そのときの吸光度を測定し、あらかじめ作成した検量線から、水銀量を求める。
全操作にわたつて空試験を行い、結果を補正する。
検量線の作成
水銀標準溶液を段階的に(総水銀として0.1~1.0μgに相当する量)取り、本文と同様に操作し、水銀量と吸光度との関係線を作成する。
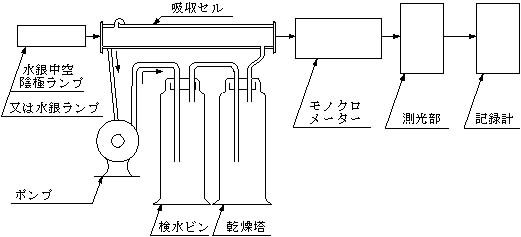
装置(装置の配置の一例を図1に示す)
光源:水銀中空陰極ランプ又は水銀ランプ
吸収セル:外径約30mm長さ100~300mmで石英製又は窓のみ石英製のもの
ポンプ:毎分1.5l以上の空気を循環できるもので通常ダイヤフラムポンプが使用される。
乾燥塔又はU字管
連結管:軟質塩化ビニル管又はポリエチレン管
図―1 原子吸光分析装置の配置の一例
(還元気化循環法)
b) 加熱気化吸引法
試験溶液の適量(総水銀として0.5~5.0μgに相当する量)を取り、塩酸ヒドロキシルアミン溶液(20W/V%)1ml及び硫酸(1+1)5mlを加え混合する。これにジチゾン―四塩化炭素溶液(0.005W/V%)10mlを加え、5分間激しく振とうする。四塩化炭素層を分離し、水層に再びジチゾン―四塩化炭素溶液(0.005W/V%)10mlを加えて振とうする。四塩化炭素層を合わせ、これに四塩化炭素を加えて正確に25mlとする。この溶液5mlを磁器ボートに移し入れ、通風して有機溶媒を揮散させた後、原子吸光分析装置の加熱部にそう入し、吸引を開始すると同時に、加熱部を加熱する。水銀ジチゾン錯塩の分解にともなつて発生する水銀蒸気を一定の速度で吸引しながら吸光度を測定する。その測定値が減少し始めるまで吸引を続け、吸光度の最大値を求め、あらかじめ作成した検量線から水銀量を求める。
全操作にわたつて空試験を行い、結果を補正する。
検量線の作成
水銀標準溶液を段階的に(総水銀として0.5~5.0μgに相当する量)取り本文と同様に操作し、水銀量と吸光度との関係線を作成する。
装置(装置の配置の一例を図2に示す)
光源:水銀中空陰極ランプ又は水銀ランプ
吸収セル:外径約30mm、長さ100~300mmで石英製又は窓のみ石英製のもの
図―2 原子吸光分析装置の配置の一例
(加熱気化吸引法)
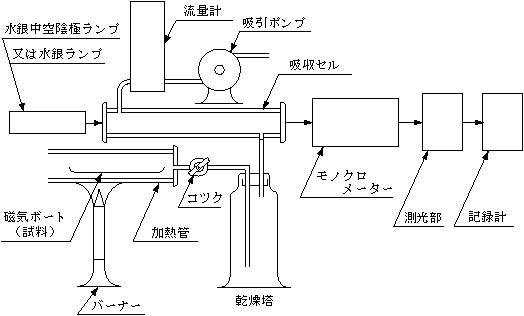
乾燥塔又はU字管
吸引装置:毎分200ml以上の空気を吸引できるもの
連結管:
軟質塩化ビニル管又はポリエチレン管
磁器ボート
加熱管
流量計
6) アルキル水銀
(1) 試薬
塩化メチル水銀標準原液又は塩化エチル水銀標準原液:塩化メチル水銀(CH3HgCl)又は塩化エチル水銀(C2H5HgCl)0.1gをベンゼンに溶かし、メスフラスコ100mlに入れ、ベンゼンを標線まで加える。この溶液1mlは、塩化メチル水銀又は塩化エチル水銀1.0mgを含む。
塩化メチル水銀標準溶液又は塩化エチル水銀標準溶液:塩化メチル水銀又は塩化エチル水銀標準原液をベンゼンを用いて適当な濃度となるように調製する。
(2) 試験溶液の調製
2―1)の分析試料約10g(湿泥)を正確にはかり取り、適当な容器に移し入れ塩酸(1+1)20ml及び硫酸銅溶液(1W/V%)5mlを加え5分間激しく振とうする。次に、ベンゼン40mlを加え5分間激しく振とうする。ベンゼン層を分離後注1)、水層に再びベンゼン40mlを加え、5分間激しく振とうする。ベンゼン層を合わせ塩化ナトリウム溶液(20W/V%)を用いて洗液が中性になるまで洗浄する。注2)洗浄後ベンゼン層にL―システイン溶液(0.1W/V%)8mlを加えて10分間激しく振とうする。水層を分離した後、水層に塩酸(1+1)2mlとベンゼン5mlを加えて5分間激しく振とうする。水層を捨てベンゼン層を無水硫酸ナトリウムで脱水後、試験溶液とする。
(3) 測定方法
ガスクロマトグラフイー条件
カラム:ガラスカラム(内径3~4mm 長さ1~2m)
充てん剤:5~25% DEGSor1.4―BDS
カラム温度:150~160℃
検出器温度:140~200℃
注入口温度:140~240℃
検出器:ECD(63Ni)
キヤリヤーガス:窒素又はヘリウム(30~80ml/min)
上記条件にセツトしたガスクロマトグラフに試験溶液をマイクロシリンジを用いて数μl注入し、ガスクロマトグラムを得る。アルキル水銀のピーク面積又はピーク高を測定し、あらかじめ作成した検量線から水銀量を求める。
測定の結果、ここに得られたピークがアルキル水銀化合物によつて得られたものであるか否かを判定するために、試験溶液の残部の1mlにグルタチオン溶液(1W/V%)1mlを加えて2分間激しく振とうし、ベンゼン層を分取し、ガスクロマトグラフに注入する。この結果、先に得られたピークの位置にピークが認められなくなつた場合には、先のピークはアルキル水銀化合物によるものと判定する。
なお、測定値は、水銀の濃度に換算しアルキル水銀態水銀で表示するものとする。
検量線の作成
L―システイン溶液(0.1W/V%)8mlに塩化メチル水銀標準溶液又は塩化エチル水銀標準溶液を段階的に取り、以下本文と同様に操作してアルキル水銀量とピーク面積又はピーク高との関係線を作成する。
- 注1) 分離し難いときは遠心分離機を用いてもよい。
- 注2) ベンゼン層に塩酸が残つていると、後のL―システイン溶液による逆抽出を妨害し、水で洗浄すると、メチル水銀が水層に移行するおそれがあるので、20%塩化ナトリウム溶液を用いる。
- 注3) 総水銀0.01ppm未満の試料についてはアルキル水銀を測定しないものとする。
7) カドミウム
(1) 試薬
ジエチルジチオカルバミン酸ナトリウム溶液(5W/V%):原子吸光用ジエチルジチオカルバミン酸ナトリウム〔(C2H5)2NCSSNa・3H2O〕6.5gを水に溶かして100mlとし、着色びんに保存する。2週間ごとに調製する。
カドミウム標準原液:金属カドミウム(99.9%以上)0.100gを硝酸(1+10)50mlに溶かし、煮沸して酸化窒素ガスを追い出し、冷却後メスフラスコ1lに入れ、水を標線まで加える。この溶酸1mlは、カドミウム0.1mgを含む。
カドミウム標準:カドミウム標準原液を水を用いて適当な濃度となるように調製する。
(2) 試験溶液の調製
2―1)の分析試料約10g(湿泥)を正確にビーカーにはかり取り、これに硝酸10mlと塩酸20mlを加え、時計ざらでフタをして注1)、熱板上で静かに加熱分解する。液量が約半分になつてから硝酸10mlを加え、再び加熱を続ける。液量が約20mlになつたらビーカーを熱板からおろして冷却する。ビーカーの壁を少量の水で洗浄し水50mlを加えて静かに加熱する不溶解物が沈降するのをまつて、ろ紙(5種B)でろ過する。ビーカーの中の不溶解物及びろ紙を少量の温塩酸(1+10)で洗浄する。ろ洗液を合わせて室温まで冷却し、水を加えて定容とし、これを試験溶液とする。
備考) 低温灰化装置を用いても差支えない。その時は2―3)で乾燥した試料を粉砕し、約2gを正確に石灰用のさらにはかり取り、低温灰化装置を用いて灰化する。
灰化が終つたら塩酸(1+10)30mlを用いて溶解し、ろ紙(5種B)でろ過する。石灰用のさら及びろ紙を少量の温塩酸(1+10)で洗浄する。ろ洗液を合わせて室温まで冷却し、水を加えて定容とし試験溶液とする。
(3) 測定方法
試験溶液の適量(カドミウムとして0.05~5μgに相当する量)にクエン酸二アンモニウム溶液(20W/V%)10mlを加え、アンモニア水(1+1)を用いてpH9~9.5に調節する。
この液にジエチルジチオカルバミン酸ナトリウム溶液(5W/V%)5mlを加え水で50mlとして後酢酸n―ブチルを正確に10ml加え激しく振とうする。酢酸n―ブチル層を分離し原子吸光光度計で吸光度を測定し、あらかじめ作成した検量線からカドミウム量を求める。
全操作にわたつて空試験を行い、結果を補正する。
検量線の作成
カドミウム標準溶液を段階的に(カドミウムとして0.05~5μgに相当する量)取り、本文と同様に操作して、カドミウム量と吸光度との関係線を作成する。
注1) 時計ざらをガラス棒を用いるなど適当な方法でうかしておく。
8) 鉛
(1) 試薬
ジチエルジチオカルバミン酸ナトリウム溶液(5W/V%):原子吸光用ジエチルジチオカルバミン酸ナトリウム〔(C2H5)2NCSSNa・3H2O)6.5gを水に溶かして100mlとし、着色びんに保存する。2週間ごとに調製する。
鉛標準原液:硝酸鉛1.60gを少量の水に溶かし、硝酸(1+1)1mlを加え、メスフラスコ1lに入れ、水を標線まで加える。この溶液1mlは鉛1mgを含む。
鉛標準溶液:鉛標準原液を水を用いて適当な濃度となるように調製する。
(2) 試験溶液の調製
2―7)、(2)のカドミウムの試験溶液の調製に準ずる。
(3) 測定方法
試験溶液の適量(鉛として0.5~50μgに相当する量)を取り、以下2―7)、(3)のカドミウムの測定方法に準じて操作を行い吸光度を測定し、あらかじめ作成した検量線から鉛量を求める。
全操作にわたつて空試験を行い結果を補正する。
検量線の作成
鉛標準溶液を段階的に(鉛として0.5~50μgに相当する量)取り本文と同様に操作して鉛量と吸光度との関係線を作成する。
9) 総クロム
(1) 試薬
亜硝酸ナトリウム溶液(10W/V%):亜硝酸ナトリウム(NaNO2)10gを水に溶かして100mlとする。この溶液は使用時に調製する。
ジフエニルカルバジドのアセトン溶液(1W/V%):ジフエニルカルバジド〔(C6H5NHNH)2CO〕0.5gをアセトン(CH3COCH3)25mlに溶かし水を加えて50mlとする。この溶液は使用時に調製する。
トリオクチルアミン・メチルイソブチルケトン溶液(TOA・MIBK):TOA{〔CH3(CH2)7〕3N}3mlをMIBK〔(CH3)2CHCH2COCH3〕に溶かして100mlとする。
クロム標準原液:重クロム酸カリウム(K2Cr2O7)(標準試薬)0.283gを水に溶かしメスフラスコ100mlに入れ、水を標線まで加える。この溶液1mlはクロム1,000μgを含む。
クロム標準溶液:クロム標準原液を水を用いて適当な濃度となるように調製する。
(2) 試験溶液の調製
2―3)の分析試料(乾燥)をメノウ製乳鉢を用いて約74μ(200mesh)以下にすりつぶし、その0.5gを磁製るつぼに取り電気炉で徐々に温度を上げ550℃で2時間灰化する。
灰化したのち、白金るつぼ又は白金ざら注1)に移し入れる。これに硫酸(1+3)数滴とふつ化水素酸20mlを加え、ドラフト内において熱板上できわめて徐々に硫酸白煙が発生し始めるまで加熱し、いつたん放冷した後、ふつ化水素酸5mlを加え硫酸白煙が発生しなくなるまで加熱する。引き続き白金るつぼを直火で徐々に温度を上げ、硫酸白煙が発生しなくなるまで加熱し放冷する。炭酸ナトリウム5g及び硝酸ナトリウム0.3gを加えふたをしたのち、るつぼを直火で徐々に温度をあげ、約900℃で、時々るつぼをゆり動かし、内容物をよく混ぜ合せ、約20分間加熱融解する。放冷時、るつぼは少量の温水を加え融成物をビーカー(200ml)に移し入れる。
ビーカーを水浴上で加温してクロム酸塩を浸出する。これをろ紙(5種B)を用いてろ過し、温水で洗浄する。ろ洗液を合わせ、硫酸(1+2)を加えて中和する。これを蒸発し、冷却後メスフラスコ100mlに移し入れ、水を標線まで加え、試験溶液とする。
ただし、残さ中にクロム分が残留する恐れのあるときは、再び融解操作を繰り返す。
(3) 測定方法
ア) 吸光光度法
試験溶液の適量(クロムとして0.015~0.25mgに相当する量)をビーカー(100ml)にとり、これに硫酸の全量が2~3mlになるように硫酸(1+2)を加え、数分間煮沸したのち冷却し、メスフラスコ50mlに洗い移し、水を標線まで加える。この溶液から正しく20mlづつをA・B2個のビーカー(100ml)に分取する。
ビーカー(A)に過マンガン酸カリウム溶液(3W/V%)を液の色が赤紫色になるまで滴下し、更に2~3滴加えたのち静かに数分間煮沸してクロムを完全に酸化する。(加熱中に液の赤紫色が消えそうになつたら過マンガン酸カリウム溶液を滴下し常時液の色を赤紫色にしておく。)
室温まで冷却した後尿素溶液(20W/V%)10mlを加え激しくかき混ぜながら亜硝酸ナトリウム溶液(10W/V%)を液の色の赤紫が消えるまで1滴づつ加える。(過剰の添加によりクロム(6価)の一部が還元される恐れがあるので注意すること。)少量の水でメスフラスコ50mlに移し入れ、過剰の亜硝酸と尿素の反応による泡が消えるまで振り混ぜる。20℃以下に冷却したのちジフエニルカルバジドのアセトン溶液(1W/V%)2mlを加え、直ちに振り混ぜ、水で標線まで希釈し、更に振り混ぜて発色させる。室温で約10分間放置後この溶液の一部を吸収セル(10mm)にとる。
ビーカー(B)の中の溶液にエチルアルコール(95W/V%)数滴を加え数分間加熱してクロムを還元する。室温まで冷却し、尿素溶液(20W/V%)10ml及び亜硝酸ナトリウム溶液(10W/V%)2~3滴を加えたのち、メスフラスコ50mlに移し入れ、過剰の亜硝酸と尿素の反応による泡が消えるまで振り混ぜ、20℃以下に冷却したのち、ジフエニルカルバジドのアセトン溶液(1W/V%)2mlを加え、直ちに振り混ぜ水で標線まで希釈し、よく振り混ぜる。室温で約10分間放置後これを対照液として吸収セルにとり、A液の吸光度を波長540nm付近で吸光度を測定し、あらかじめ作成した検量線からクロム量を求める。
検量線の作成
クロム標準溶液を段階的に(クロムとして0.015~0.25mgに相当する量)取り硫酸(1+2)2mlを加え、数分間煮沸した後冷却し、メスフラスコ50mlに洗い移し、水を標線まで加える。以下本文に従つて操作し、クロム量と吸光度との関係線を作成する。
イ) 原子吸光光度法
試験溶液の適量(クロムとして10~100μgに相当する量)をとり、水を加えて100mlとする。これに硫酸(3+50)を用いて中和する。これに塩析剤として硫酸アンモニウム溶液(40W/V%)20ml、硫酸(3+50)20ml及び水を加えて200mlとし、更にTOA・MIBK溶液20mlを加えて10分間振り混ぜた後、MIBK層をとり、この液を原子吸光光度計を用いて吸光度を求め、別に作製した検量線より試料中のクロムの濃度を求める。全操作にわたつて空試験を行い結果を補正する。
検量線の作成
クロム標準溶液を段階的に(クロムとして10~100μgに相当する量)取り、本文と同様に操作してクロム量と吸光度との関係線を作成する。
- 注1) 白金るつぼ又は白金ざらがない場合は、次の方法で行う。
2―3)の乾燥試料を約74μ(200mesh)以下にすりつぶし、その0.5gをはかり取り、ニツケルるつぼ又は鉄るつぼに入れ、電気炉で徐々に温度を上げ550℃で2時間灰化する。
冷却後過酸化ナトリウム約5gを入れ混合し、徐々に温度を上げて加熱し、約5分間赤熱を持続して融解した後冷却する。これをるつぼのまま50mlの水を加えたビーカー(容量300ml)に移し入れる。
(激しい反応に注意すること)。温水約50mlを注意しながら少しづつ加え冷却する。
るつぼを水で洗つてとり出す。浸出液に過酸化ナトリウム0.1gを少しづつかき混ぜながら加え加熱煮沸してクロムを酸化し、更に煮沸を続けて過剰の過酸化ナトリウムを分解する。これを20℃以下に冷却したのち250mlのメスフラスコに移し入れ、水で標線まで薄め振り混ぜる。しばらく静置した後ろ紙(5種B)を用いてろ過し、初めのろ液約50mlは捨て、次のろ液から100mlを試験溶液とする。
10) 6価クロム
(1) 試薬
ジフエニルカルバジドのアセトン溶液(1W/V%):ジフエニルカルバジド〔(C6H5NHNH)2CO〕0.5gアセトン25mlに溶かし水を加えて50mlとする。この溶液は使用時調製する。
トリオクチルアミン・メチルイソブチルケトン溶液(TOA・MIBK):TOA{〔CH3(CH2)7〕3N}3mlをMIBK〔(CH3)2CHCH2COCH3〕に溶かして100mlとする。
クロム標準原液:重クロム酸カリウム(K2Cr2O7)(標準試薬)0.283gを水に溶かし、メスフラスコ100mlに入れ、水を標線まで加える。この溶液1mlはクロム1,000μgを含む。
クロム標準溶液:クロム標準原液を水を用いて適当な濃度となるように調製する。
(2) 試験溶液の調製
混合液中に含まれる乾燥固形分と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるように2―1)の分析試料(湿泥)を取り、水を加えて混合液を調製し、室温において4時間連続して、かくはん又は振とうする。
約30分放置した後、ろ紙(5種C)を用いてろ過し定容とし、試験溶液とする。
(3) 測定方法
ア) 吸光光度法
試験溶液の適量〔クロムとして0.015~0.25mgに相当する量〕をメスフラスコ50mlに取り、硫酸(1+1)0.5~0.6mlを加えて振り混ぜ、液温を15℃に冷却する。これにジフエニルカリバジドのアセトン溶液(1W/V%)0.5mlを加え直ちに振りまぜ、水を標線まで加えて振り混ぜ、10分後にその一部を吸収セル(10mm)に移す。別のビーカー(100ml)に先に分取した検水の同量を取り、硫酸(1+1)0.5~0.6mlを加え、エチルアルコール(95V/V%)少量を加え、煮沸してクロム酸を還元する。液温を15℃に冷却し、メスフラスコ50mlに移し入れ、これにジフエニルカルバジドのアセトン溶液(1W/V%)0.5mlと水を標線まで加えて振り混ぜ、10分後にその一部を吸収セル(10mm)に移す。
この溶液を対照液として先に発色させた溶液の吸光度を波長540nm付近で測定し、あらかじめ作成した検量線からクロム量を求める。
検量線の作成
クロム標準溶液を段階的に(クロムとして0.015~0.25mgに相当する量)取り、硫酸(1+1)0.5~0.6mlを加え、以下本文と同様に操作し、クロム量と吸光度との関係線を作成する。
イ) 原子吸光光度法
試験溶液の適量(クロムとして10~100μgに相当する量)を取り、水を加えて100mlとする。これに硫酸(3+50)を加えてpHメーターを用いて中和する。これに塩析剤として硫酸アンモニウム溶液(40W/V%)20ml、硫酸(3+50)20ml及び水を加えて200mlとし、更にTOA・MIBK溶液20mlを加えて10分間振り混ぜた後、MIBK層を取り、この液を原子吸光光度計を用いて吸光度を求め、別に作製した検量線より試料中のクロムの濃度を求める。全操作にわたつて空試験を行い結果を補正する。
検量線の作成
クロム標準溶液を段階的に(クロムとして10~100μgに相当する量)取り、本文と同様に操作してクロム量を吸光度との関係線を作成する。
- 注1) この方法では、全6価クロムが抽出されない場合もあるが、現時点では、他によい方法がないのでこの方法で行うこと。
- 注2) この方法は、クロムの溶出試験とほぼ同じであるので、あらためて溶出試験を行う必要はない。
11) ひ素
(1) 試薬
塩化第一すず溶液:塩化第一すず(SnCl2・2H2O)40gを塩酸50mlで溶かして水を加えて100mlとする。これに金属すずの小粒を加えて保存する。この溶液を10倍に薄めて使用する。
酢酸鉛溶液(10W/V%):酢酸鉛〔Pb(CH3COO)2・3H2O〕11.8gを水と酢酸1~2滴に溶かし、水で100mlとする。
砂状亜鉛(1000~1410μ):無ひ素亜鉛(ひ素0.1ppm以下のもの)を塩酸(1+10)と水で表面を洗い乾燥した後使用する。
ジエチルジチオカルバミン酸銀のブルシンクロロホルム溶液:ジエチルジチオカルバミン酸銀〔(C2H5)2NCSSAg〕0.25gとブルシン(C23H26O4N2・2H2O)0.10gをクロロホルム(CHCl3)に溶かし正確に100mlとし、暗所に保存する。
ひ素標準原液:三酸化ひ素(As2O3)0.133gをメスフラスコ100mlに取り、水酸化ナトリウム溶液(4W/V%)2mlに溶かし水で薄め、硫酸(1+10)で微酸性とし、水を標線まで加える。この溶液1mlは、ひ素1mgを含む。
ひ素標準溶液:ひ素標準原液を水を用いて適当な濃度となるように調製する。
(2) 試験溶液の調製
2―1)の分析試料約10g(湿泥)を正確にはかり取り、ケールダールフラスコに移し、水約50mlを加えて懸濁させた後、硝酸約15ml、硫酸約15mlを加え、徐々に加熱して液量が約50mlとなつたら強熱し、硫酸の白煙が生じたら直に加熱をやめ、硝酸10mlを追加して再び加熱し、硫酸の白煙が生じたら直ちに加熱をやめる。この操作を繰り返し、有機物を分解する。放冷後ガラス綿又はガラス繊維ろ紙でろ過し、ろ紙を水で洗浄する。ろ洗液を合わせ溶液を冷却し定容として試験溶液とする。
(3) 測定方法
試験溶液の適量(ひ素として1~40μgに相当する量)をひ化水素発生びん(図―3)に入れ、水を加えて約40mlとする。次にヨウ化カリウム溶液(20W/V%)15mlと塩化第一すず溶液5mlを加え、混合し、約1分間室温に放置する。発生びんAに砂状亜鉛5gを加え、手早く発生びんA、導管B及びジエチルジチオカルバミン酸銀のブルシンクロロホルム溶液5mlを入れた吸収管Cを連結し、25℃の水中で約1時間放置し発生したひ化水素を吸収し、発色させる。これにクロロホルムを加え、正確に5mlとし、1部を吸収セル(10mm)に取り、ジエチルジチオカルバミン酸銀のブルシンクロロホルム溶液を対照液として波長530nm付近での吸光度を測定しあらかじめ作成した検量線からひ素量を求める。
全操作にわたつて空試験を行い結果を補正する。
検量線の作成
ひ素標準溶液を段階的に(ひ素として1~40μgに相当する量)発生びんに取り、本文と同様に操作してひ素量と吸光度との関係線を作成する。
図―3 ひ化水素発生器及び吸収管の一例
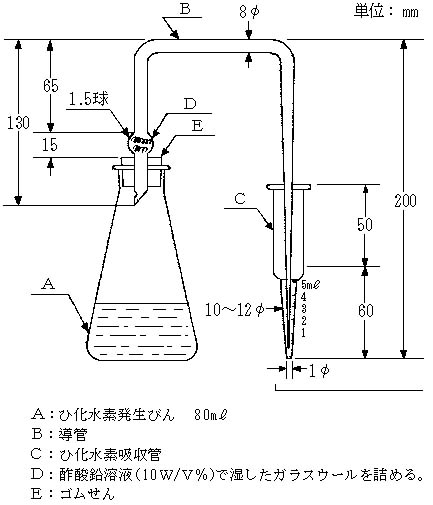
- A:ひ化水素発生びん 80ml
- B:導管
- C:ひ化水素吸収管
- D:酢酸鉛溶液(10W/V%)で湿したガラスウールを詰める。
- E:ゴムせん
12) BHC
(1) 試薬
フロリジル:アルミニウム製又は磁製のさらに入れ、130℃で15時間加熱する。加熱後デシケーター内で放冷する。フロリジルの性能は、ロツト毎に若干変動するので予めBHCの溶出範囲を調べておく。
BHC標準原液:BHC4異性体の各々の純品をn―ヘキサンを用いて、各々100ppm溶液とする。
BHC標準溶液:BHC標準原液をn―ヘキサンを用いて適当な濃度となるように調製する。なお、4異性体を混合して調製してもよい。
(2) 試験溶液の調製
2―1)の分析試料約20g(湿泥)を共せん付フラスコに取り、アセトン50mlを加え30分間振とうする。振とう後しばらく放置して、ろ紙(5種C)でろ過する。ろ紙上の残さを少量の水とアセトン50mlを用いてフラスコ内に洗い入れる。再度30分間振とうしろ過する。ろ液を合わせて約30mlまでクデルナーダニツシユ濃縮器(ロータリーエバポレーターを使用してもよい)を用いて濃縮する。濃縮液に塩化ナトリウム溶液(2W/V%)300ml、n―ヘキサン100mlを加えて激しく5分間振とうする。ヘキサン層を分離し水層に再びn―ヘキサン100mlを加えて激しく5分間振とうする。ヘキサン層を分離し、先のヘキサンと合わせ100mlの水で2回洗浄する。無水硫酸ナトリウムを用いて脱水し、ヘキサン層をデカンテーシヨン又はろ紙(5種B)でろ過する。少量のn―ヘキサンで無水硫酸ナトリウムを洗浄し、ろ洗液を合わせる。
ヘキサンをクデルナーダニツシユ濃縮器を用いて約5mlまで濃縮しフロリジルカラムクロマト試料とする。
フロリジルカラムクロマトグラフイー
10×300mmのカラムクロマト用ガラス管の底部に脱脂綿又は、ガラスウールをつめ、n―ヘキサンで洗浄する。
フロリジル5gにn―ヘキサンを加え撹拌し気泡を除きカラムに充てんしn―ヘキサン10mlを流下させる。無水硫酸ナトリウム1gを上積した後、前記濃縮液を静かに流下させ少量のn―ヘキサンを用いてガラス壁を洗う。6%エーテル含有n―ヘキサン100mlを用いて溶出を行い、これを濃縮して試験溶液とする。
(3) 測定方法
ガスクロマトグラフイー条件
カラム:ガラスカラム(内径3~4mm、長さ1~2m)検量線の作成
充てん剤:5%、OV―17
カラム温度:150~200℃
検出器温度:180~250℃
注入口温度:180~250℃
検出器:ECD(63Ni)
キヤリヤーガス:窒素又はヘリウム50~100ml/min
上記条件にセツトしたガスクロマトグラフに(2)で調製した試験溶液をマイクロシリンジを用いて数μl注入しガスクロマトグラムを得る。BHCの4異性体の各ピーク面積を測定し、あらかじめ作成した検量線からBHC量を求める。
BHCの標準溶液を段階的にガスクロマトグラフに注入し、ガスクロマトグラムを得、BHCの4異性体の各ピーク面積と重量との関係線を作成する。
- 注1) PCBが共存する場合は、クロマトグラム上で、PCBの一部とBHCは重なる場合がある。この場合は次のPCBのときに行う。
「シリカゲルクロマトグラフイー」を行い分離することとする。
この分離の方法は、最初n―ヘキサンで展開しPCBのみを溶出分離し、その後、BHCを10%エーテル含有n‐ヘキサンで溶出する。
なお、シリカゲルも、フロリジルと同様に活性化等によつて溶出範囲が違つてくるのであらかじめ、溶出範囲を調べておく。 - 注2) 分子状イオウが共存し、ガスクロマトグラフによる定量を妨害する場合は、あらかじめ、次の銅チツプ処理を行う。
(銅チツプ処理)
直径1mm程度の銅線を2~3mmの長さに切断したものを用意し(10~20g)ガラス共栓付試験管(15~30ml)に入れ濃塩酸15mlを加えて激しく振とうする。
傾斜して塩酸を除去した後、蒸留水を加えて水洗し、塩酸を十分除去した後、アセトンで水を置換し、ドライヤーで手早く乾燥する。
この銅のチツプはn―ヘキサン中に保存する(使用後表面の黒化した銅チツプもこの方法で再生する。)
試料溶液をガラス共栓付試験管に取り銅チツプ約5gを加えて約30秒激しく振とうする。
しばらく静置後、上澄み液をコマゴメピペツトで注意深く別の共栓試験管に移し、再び数粒の銅チツプを加え、軽く振りまぜる。これで銅チツプが黒変しなければガスクロマトグラフ測定に供する。
黒変した場合は、再び5g程度の銅チツプを加えて振とうする。
以下同様にして銅チツプの表面に黒変がなくなるまで反復処理する。
13) ポリクロリネイテツドビフエニル(PCB)
(1) 試薬
n―ヘキサン:ガスクロマトグラフに注入(300mlを約3mlに濃縮し、その10μlを分取して注入する)したとき、PCBの保持時間にピークを生じないもの。
水酸化カリウムのエチルアルコール溶液:水酸化カリウム(KOH)70gをできるだけ少量の水(水1lにつきn―ヘキサン100mlを用いて振り混ぜ洗浄したもの、以下同じ。)に溶かし、エチルアルコール(C2H5OH)(95V/V%)注1)を加えて1lとして振とうし、二酸化炭素に触れないようにして2~3日放置したのち、その上澄み液を取り、(又はろ過する。)耐アルカリ性びんに保存する。
無水硫酸ナトリウム:硫酸ナトリウム(Na2SO4)(無水)100gにn―ヘキサン50mlを加えて振とうし、ろ別する。残分にn‐ヘキサン25mlを加えて振とうし、ろ別したのち風乾する。この場合後者のろ別したn‐ヘキサン10μlをガスクロマトグラフに注入したとき、PCBの保持時間にピークを生じないものを用いる。
シリガゲル:PCB分析用シリカゲル粉末をビーカーに入れ、層の厚さを10mm以下にして約130℃で18時間以上乾燥したのち、デシケーター中で約30分間放冷する。この場合シリカゲルクロマト管によるPCBの分離の操作の空試験を行い、その試験溶液10μlを分取してガスクロマトグラフに注入したとき、PCBの保持時間にピークを生じないものを用いる。
PCB標準液:試験用PCBのKC―300、KC―400、KC―500及びKC―600を重量比1:1:1:1の割合で混合したものをn―ヘキサンを用いて適当な濃度となるように調製する。
(2) 試験溶液の調製
2―1)の分析試料約10g(湿泥)を正確にはかり取り、これを還流冷却器付フラスコに入れ、水酸化カリウムのエチルアルコール溶液50mlを加え約1時間沸騰湯浴上で加熱分解を行い、約50℃まで放冷し注2)、n―ヘキサン50mlを加えて振とうする。静置して室温まで放冷したのち、ガラスウールを敷いたロート注3)を用いてろ過し分液ロートに入れる。フラスコの内容物をn―ヘキサン20mlずつで3回洗い洗液をろ過して分液ロートに合わせる。試料中の水分を考慮して水の総量が約50mlになるように水を加える。ゆるやかに振とうした後n―ヘキサンが十分分離するまで静置し注4)、n―ヘキサン層を分離し、水層を他の分液ロートに入れ再びn―ヘキサン50mlを加え同様に抽出を行い、n―ヘキサン層を先のn―ヘキサン層と合わせる。更にn―ヘキサン層を水100mlずつでゆるやかに振とうし3回洗浄する。n―ヘキサン層を無水硫酸ナトリウム約10gを用いて脱水したのち、全量が約5mlになるまで濃縮する。
シリカゲルカラムクロマトグラフイー注5)10×300mmのカラムクロマト用ガラス管の底部に脱脂綿又はガラスウールをつめ、これにn―ヘキサンを加えて完全に気泡を除去する。シリカゲル4gを容器に取り、n―ヘキサンを加えて気泡を除去した後、クロマト管に充てんする。
無水硫酸ナトリウム1gを上積した後、濃縮液を静かにクロマト管に入れn―ヘキサン少量を用いて濃縮液の入つていた容器及びクロマト管内壁を洗う。濃縮液を無水硫酸ナトリウム層の面まで流下させる。n―ヘキサン500mlを入れた分液ロートを装着し、毎秒1滴程度の流下速度で注6)全PCBが含まれかつ、PCB及びDDE以外の有機塩素化合物が含まれないような流出範囲注7)で、クロマト管からの流出液を容器に集める。流出液の全量を5ml以下になるまで濃縮し、n―ヘキサンを加えて正確に5mlとし、試験溶液とする。
(3) 測定方法
ガスクロマトグラフイー条件
カラム:ガラスカラム(内径2~4mm、長さ1.5~2m)
充てん剤:酸洗浄後シラン処理したガスクロムQ、クロモソルブG又はクロモソルブW(149~177μ)にOV―1又はOV―17を1.5~5%被覆したもの
カラム温度:180~250℃
検出器温度:200~250℃
注入口温度:200~250℃
検出器:ECD
キヤリヤーガス:窒素又はヘリウム30~80ml/min
上記条件にセツトしたガスクロマトグラフにマイクロシリンジを用いてPCB標準液5μlを注入し、得られたクロマトグラムのピークに別図を参考にして番号を付ける。次にそのピークごとにピークの高さを読みとり、その高さ(H1)と当該ピークのピーク番号に対応する別表のCB0(%)から次式によりK値を求める。
K=CB0(%)/H1
次に試験溶液1~10μlを同様にガスクロマトグラフに注入し、得られたクロマトグラムのピークにその位置に相当するPCB標準液で得られたクロマトグラムの位置のピークのピーク番号と同一のピーク番号を付ける。次にそのピークごとに、ピークの高さを読みとり、その高さ(H2)と当該ピークのピーク番号に係るK値から次式によりCB2(%)を求める。
CB2(%)=K×H2
以上の結果から、次式により、試料中のPCB濃度を求める。
PCB濃度=PCB標準液のPCB濃度×(PCB標準液注入量/試験溶液注入量)×(ΣCB2(%)/ΣCB0(%))×(試験溶液量/試料採取量)
- 注1) n―ヘキサンと同様にして、ガスクロマトグラフに注入したときピークを生じないもの。
- 注2) 温度が高いときに、n―ヘキサンを加えると、沸騰するので危険である。また、あまり冷却しすぎるとアルカリ分解によつて生成したケン化物がn―ヘキサンに溶けにくくなり、PCB抽出のロスの原因となる。
- 注3) ガラス繊維ろ紙を用いてもよいが、目づまりを起こしやすくろ過が困難になることが多い。
- 注4) エマルジヨンを生じる場合は、エチルアルコール数mlを加え、緩やかに振り混ぜる。
- 注5) 油分等を多く含む底質で、アルカリ分解法によつても分解されず、多量に油分等の残る試料については、シルカゲルクロマトグラフイーを行う前に、アセトニトリル・n―ヘキサン分配法により油分等を除去する必要がある。
アセトニトリル・n―ヘキサン分配法
濃縮液を少量のn―ヘキサンを用いて100mlの分液ロートに洗い込み、(総n―ヘキサン量は約15mlとする)n―ヘキサン飽和アセトニトリル(n―ヘキサン約30mlとアセトニトリル約100mlを分液ロートに入れ振とう後アセトニトリル層を使用する。)30mlを加え約1分間振とう抽出したのち、分離するまで静置する。アセトニトリル層を分離し、n―ヘキサン層をn―ヘキサン飽和アセトニトリル30mlずつで更に3回抽出する。全アセトニトリル層を、塩化ナトリウム溶液(2W/V%)700ml及びn―ヘキサン100mlを入れた分液ロート中に移し、緩やかに振り混ぜ十分分離するまで静置する。次に水層を別の分液ロートに移し、これにn―ヘキサン100mlを加え、激しく振とうし、分離するまで静置する。水層を捨て、n―ヘキサン層を合わし、塩化ナトリウム(2%)100mlずつで2回洗浄する。n―ヘキサン層を無水硫酸ナトリウム10gを用いて脱水後約5mlになるまで濃縮する - 注6) 必要があれば窒素ガスで加圧する。
- 注7) この流出範囲は、試料中のPCBの含有量、シリカゲルの活性度のわずかな差異等により変動するので、あらかじめ試験用PCBを用いてカラムクロマトグラフ操作を数回行いPCBの流出範囲とその安定性を十分確認しておく必要がある。
(1) 分離管充てん物の被覆にOV―1を用いたときのクロマトグラム
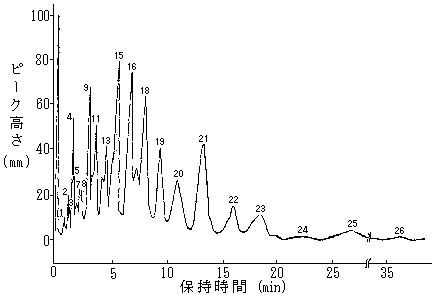
(2) 分離管充てん物の被覆にOV―17を用いたときのクロマトグラム
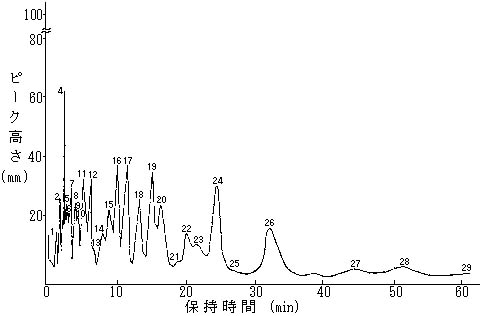
(3) ポリクロリネイテツドビフエニル試験成績表(例)
カラムOV―1
|
塩化物
|
Cl2
|
Cl3
|
Cl4
|
Cl5
|
Cl6
|
Cl7
|
Cl8
|
Cl9
|
TotalCB0(%)
|
Calc
|
Total
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
ピークNO
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
13
|
14
|
15
|
16
|
17
|
18
|
19
|
20
|
21
|
22
|
23
|
24
|
25
|
26
|
PCBppm
| ||||
| \ |
K
|
0.084
|
0.231
|
0.122
|
0.085
|
0.122
|
0.239
|
0.107
|
0.063
|
0.098
|
0.049
|
0.068
|
0.077
|
0.063
|
0.023
|
0.046
|
0.048
|
0.029
|
0.040
|
0.051
|
0.072
|
0.046
|
0.049
|
0.091
|
0.073
|
0.079
|
0.105
|
||||
|
試料
|
\ | ||||||||||||||||||||||||||||||
|
標準液KC300、400、500、600(1:1:1:1)1ppm、5μl
|
CB0
|
1.67
|
5.78
|
2.68
|
7.57
|
5.23
|
7.88
|
4.83
|
3.30
|
10.68
|
2.37
|
5.70
|
3.16
|
4.20
|
1.24
|
6.44
|
6.16
|
1.68
|
4.45
|
3.45
|
3.15
|
3.47
|
1.27
|
1.54
|
0.29
|
0.71
|
0.21
|
100.05
|
1×5/10×25.8/100.05×5/2=0.32
|
0.32ppm
|
|
|
H1(mm)
|
20
|
25
|
22
|
89
|
43
|
33
|
45
|
52
|
109
|
48
|
84
|
41
|
67
|
54
|
140
|
129
|
57
|
111
|
67
|
44
|
75
|
26
|
17
|
4
|
9
|
2
|
|||||
|
試料2g→5ml10μl
|
H2
|
3
|
9
|
2
|
16
|
18
|
17
|
33
|
36
|
11
|
16
|
20
|
11
|
64
|
50
|
10
|
37
|
12
|
4
|
10
|
2
|
2
|
25.8
|
||||||||
|
CB2(%)
|
0.25
|
0.77
|
0.24
|
3.82
|
1.93
|
1.07
|
3.23
|
0.54
|
2.45
|
1.23
|
1.26
|
0.25
|
2.94
|
2.40
|
0.29
|
2.48
|
0.61
|
0.29
|
0.46
|
0.10
|
0.18
|
||||||||||
|
CB(%)
|
1.0
|
3.9
|
41.0
|
31.5
|
16.2
|
5.7
|
0.7
|
||||||||||||||||||||||||
備考 本表で標準5ngをガスクロマトグラフに注入してピーク番号5のピーク高Hが43mmのとき、そのピークの含有率が5.23%であるので、そのピークのトリクロリネイテツドビフエニル含量は5×5.23/100ng、高さ1mmに対しては、5ng×5.23/100×1/43mm=5/100 ×5.23/43=0.0061ngである。しかし、計算が複雑になるため5/100を省き比例数(K)で計算することとした。
この場合、1mg/lを標準としているのでTotalCB0(%)は、100.05、2mg/lを標準とした場合は2mg/lが100.05、1ppmは50.03となる。
(1) 分離管充てん物の被覆にOV―1を用いたときのCB0(%)
|
ピーク番号
|
a
|
b
|
1
|
c
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
13
|
14
|
15
|
16(注)
|
17
|
18
|
19
|
20
|
21
|
22
|
23
|
24
|
25
|
26
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
CB0(%)
|
0.53
|
0.04
|
1.67
|
0.37
|
5.78
|
2.68
|
7.57
|
5.23
|
7.88
|
4.83
|
3.30
|
10.68
|
2.37
|
5.70
|
3.16
|
4.20
|
1.24
|
6.44
|
6.16
|
1.68
|
4.45
|
3.45
|
3.15
|
3.47
|
1.27
|
1.54
|
0.29
|
0.71
|
0.21
|
ΣCB0(%)=100.05
(注) ピーク番号16は条件により、16(CB0(%)2.16)及び16(CB0(%)4.00)に分離することがある。
(2) 分離管充てん物の被覆にOV―17を用いたときのCB0(%)
|
ピーク番号
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
13
|
14
|
15
|
16
|
17
|
18
|
19
|
20
|
21
|
22
|
23
|
24
|
25
|
26
|
27
|
28
|
29
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
CB0(%)
|
1.69
|
6.00
|
3.17
|
6.60
|
2.74
|
1.35
|
8.62
|
4.86
|
2.54
|
2.09
|
8.65
|
7.05
|
0.99
|
3.18
|
5.42
|
6.35
|
4.28
|
4.00
|
4.75
|
2.82
|
0.23
|
2.26
|
1.57
|
3.30
|
0.08
|
2.95
|
0.28
|
0.71
|
0.15
|
ΣCB0(%)=98.18
3 溶出試験
1) 溶出率の算定法
溶出率は次式で求めるものとする。
溶出率=W2/W1
W1:溶出試験に使用した分析試料中の有害物質量(μg)
W2:溶出試験に使用した混合液の体積に相当する溶出水中に含まれる有害物質量(μg)
2) 総水銀
(1) 試薬
2―5)(1)の総水銀の試薬に準ずる。
(2) 試験溶液の調製
混合液中に含まれる乾燥固形分の重量と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるように分析試料(湿泥)を取り、水を加えて混合液を調製し、室温において4時間連続して、かくはん又は振とうする。
約30分放置したのち、ろ紙(5種C)を用いてろ過する。そのろ液から、100mlを還流冷却器付フラスコに取り、硝酸(1+1)50ml及び過マンガン酸カリウム溶液(6W/V%)20mlを加えて2時間加熱還流する。もしこの間に過マンガン酸カリウムの色が消失する場合は室温まで冷却した後、過マンガン酸カリウム溶液(6W/V%)5mlを追加し、再び加熱する。この操作を、赤紫色が約10分間残るまで繰返す。液温を約40℃に冷却し、溶液を振り混ぜながら、塩酸ヒドロキシルアミン溶液(20W/V%)を滴加し、過剰の過マンガン酸カリウムを分解する。溶液が微紅色になるまで過マンガン酸カリウム溶液(6W/V%)を滴加し、水を加えて定容とし、これを試験溶液とする。
(3) 測定方法
2―5)(3)a)の還元気化循環法及び2―5)(3)b)の加熱気化吸引法に準ずる。
- 注1) この試験は乾燥固形分当たり総水銀含有量10ppm以上のものについて適用する。
3) アルキル水銀
(1) 試薬
2―6)(1)のアルキル水銀の試薬に準ずる。
(2) 試験溶液の調製
混合液中に含まれる乾燥固形分の重量と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるように分析試料(湿泥)を取り、水を加えて混合液を調製し、室温において4時間連続して、かくはん又は振とうする。
約30分放置したのち、ろ紙(5種C)を用いてろ過する。そのろ液から100mlを分液ロートにとりアンモニア水又は塩酸で中和したのち塩酸を加えて0.5N塩酸酸性とする。
ベルゼン50mlを加え、5分間激しく振とうする。
ベンゼン層を分離後水層に再びベンゼン50mlを加え、5分間激しく振とうする。
ベンゼン層を合わせる。以下、2―6)(2)のアルキル水銀の試験溶液の調製に準ずる。
(3) 測定方法
2―6)(3)のアルキル水銀の測定方法に準ずる。
4) カドミウム
(1) 試薬
2―7)(1)のカドミウムの試薬に準ずる。
(2) 試験溶液の調製
混合液中に含まれる固形分の重量と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるように分析試料をとり、水を加えて混合液を調製し、室温において4時間連続してかくはん又は振とうする。
約30分放置したのち、ろ紙(5種C)を用いてろ過する。そのろ液から100mlをビーカーにとり、塩酸5mlを加え、熱板上で徐々に加熱する。液量が約10mlになつたら室温まで冷却し、定容として試験溶液とする。
(3) 測定方法
2―7)(3)のカドミウムの測定方法に準ずる。
5) 鉛
(1) 試薬
2―8)(1)の鉛の試薬に準ずる。
(2) 試験溶液の調製
3―4)(2)のカドミウムの試験溶液の調製に準ずる。
(3) 測定方法
2―8)(3)の鉛の測定方法に準ずる。
6) ひ素
(1) 試薬
塩化第二鉄溶液:塩化第二鉄(FeCl3・6H2O)5gを塩酸(1+1)10mlに溶かし、水で100mlとする。
塩化第一すず溶液:塩化第一すず(SnCl2・2H2O)50gを塩酸(1+1)100mlに溶かす。
メタクレゾールパープルのエチルアルコール溶液(0.05W/V%):メタクレゾールパープル〔2―HO3SC6H4C(C6H3―2―CH3―4―O)C6H3―2―CH3―4―OH〕0.05gをエチルアルコール(C2H5OH)(95V/V%)50mlに溶かし、水で100mlとする。
他の試薬は2―11)(1)のひ素の試薬に準ずる。
(2) 試験溶液の調製及び濃縮操作
混合液中に含まれる乾燥固形分の重量と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるように分析試料(湿泥)を取り、水を加えて混合液を調製し、室温において4時間連続して、かくはん又は振とうする。
約30分放置した後、ろ紙(5種C)を用いてろ過する。ろ液の適量(ひ素として0.04mg以下に相当する量)を取り、ろ液1lにつき硝酸3mlと過マンガン酸カリウム溶液(0.3W/V%)を滴加して着色させる。これを煮沸して過マンガン酸が分解して脱色されたときは、再び過マンガン酸カリウム溶液を脱色されなくなるまで加える。過酸化水素水(1+30)のできるだけ少量を加え、過剰の過マンガン酸を分解する。塩化第二鉄溶液1mlを加え約80℃の液温になつたなら、メタクレゾールパープルのエチルアルコール溶液(0.05W/V%)数滴を加える。かき混ぜながらアンモニア水(1+2)で紫になるまで中和する(このときひ素の共沈はpH9~10が最適である)。沈澱物が沈降したのち小形ろ紙でろ過する。
(3) 測定方法
(2)で得た沈澱物を、なるべく少量の水でひ化水素発生びんAに入れ、温硫酸(1+5)18mlと塩酸(1+1)2mlを加えて沈澱物を溶かし、水で約40mlとする。
以下2―11)(3)のひ素の測定方法に準ずる。
7) BHC
(1) 試薬
2―12)(1)のBHCの試薬に準ずる。
(2) 試験溶液の調製
混合液中に含まれる乾燥固形分の重量と混合液の体積との比(g/ml)が3/100になるようにし、かつ混合液量が500ml以上になるように分析試料(湿泥)を取り、水を加えて混合液を調製し、室温において4時間連続して、かくはん又は振とうする。
約30分放置したのち、ガラス繊維ろ紙を用いてろ過する。ろ液から100mlを分液ロートに取り、飽和食塩水6mlとn―ヘキサン50mlを加え、激しく5分間振とうする。ヘキサン層を分離し、水層に再びn―ヘキサン50mlを加えて激しく振とうする。ヘキサン層を分離し、先のヘキサンと合わせる。以下2―12)(2)のBHCの試験溶液の調製に準ずる。
(3) 測定方法
2―12)(3)のBHCの測定方法に準ずる。
4 その他の項目
1) 過マンガン酸カリウム(0.1N)による酸素消費量
廃水中の沈降物などのために底質中に含まれている酸化されうる有機物の量を示すために、底質を過マンガン酸カリウムによつて酸化し、その酸素消費量を過マンガン酸カリウム(0.1N)による酸素消費量と呼ぶ。各試泥は湿つたままの状態で取り、過マンガン酸カリウムで酸化するが、これをあらわすには普通試料の乾燥重量をもととし、乾泥1g当り消費される酸素のmg数であらわす。
(1) 試薬
過マンガン酸カリウム溶液(0.1N):過マンガン酸カリウム(KMnO4)3.2gを水に溶かし1lとする。この液はよく振りまぜて2~3日放置して上澄をとるか、ガラスろ過器等でろ過し、よく洗つた褐色ビンにたくわえる。
ヨウ素酸カリウム溶液(0.1N):ヨウ素酸カリウム(KIO3)を120~140℃で2時間乾燥し、デシケータ中で放冷した後、ヨウ素酸カリウム100%に対し0.891gを正しくはかり取り、水に溶かしてメスフラスコ250mlに入れ、水を標線まで加える。
シユウ酸標準溶液(0.1N):シユウ酸(C2H2O4・2H2O)6.304gを水に溶かして正確に1lにする。
チオ硫酸ナトリウム溶液(0.1N):チオ硫酸ナトリウム(CNa2S2O3・5H2O)2.5gを水に溶かして1lとする。これに約0.1gの炭酸ナトリウムを安定剤として加える。この溶液の標定は、ヨウ素酸カリウム標準溶液(0.1N)を用いて行う。ただし、ここでは0.1Nヨウ素酸カリウム溶液をそのまま希釈せずに滴定に用いる。
水酸化ナトリウム溶液:水酸化ナトリウム(NaOH)300gを1lの水に溶かす。
ヨウ化カリウム溶液:特級ヨウ化カリウム(KI)25gを250mlの水に溶かし、褐色ビンに入れ暗所に保存する。溶液に色の付いたものを用いてはならない。
澱粉溶液:可溶性澱粉1gを小量の水で粥状に練り、100mlの沸騰した水に加え、透明になるまで煮た後、冷却して使用する。100mlの澱粉溶液に0.1gの安息香酸を加えておけばある程度の貯蔵は可能である。
(2) 試験操作
2―1)の分析試料10g(湿泥)をはかり取り、容量の300mlの三角フラスコに移し、過マンガン酸カリウム溶液(0.1N)を正確に100ml、水酸化ナトリウム溶液5mlを加え、沸騰している水浴中に15分間保つ。加熱後直ちにシユウ酸標準溶液(0.1N)を正確に100ml及び硫酸10mlを加えて過マンガン酸の色を褪色させ、室温にまで冷却する。三角フラスコの内容物を500mlの定容フラスコに水を用いて完全に移して定容とし、十分振りまぜた後、乾燥ろ紙を用いてろ過する。ろ液100mlを三角フラスコに取り、これに過マンガン酸カリウム溶液(0.1N)10mlを加えて、かき混ぜながら数分間放置し、ヨウ化カリウム溶液5mlを加えてヨウ素を遊離させた後チオ硫酸ナトリウム溶液(0.1N)で澱粉溶液を指示薬として滴定する。あらかじめ水50mlを取り、これについて上に述べたと同じ操作で空試験を行つておく必要がある。
〔結果の計算と表示〕
チオ硫酸ナトリウム溶液(0.1N)1mlは0.8mgの酸素に相当する。チオ硫酸ナトリウム溶液の滴定値を(t)ml、空試験値を(b)ml、その力価をfNa2S2O3、乾燥試料の湿試料に対する重量百分率を(D)%とすれば、乾泥1gに対する酸素消費量は次のように求められる。
酸素消費量(O2mg/1g乾泥)=(0.8×{(d)-(t)}×5×fNa2S2O3×100/10×(D))
- 注1) 過マンガン酸カリウム溶液(0.1N)とシユウ酸標準溶液(0.1N)の相対濃度はシユウ酸溶液の方をやや濃くしておくことが必要である。
- 注2) 滴定を始める前に過マンガン酸カリウム溶液(0.1N)10mlを加えても全部が消費されて色が消失した場合には更に10mlを追加する。この場合は当然空試験にも20mlの過マンガン酸カリウム溶液を用いねばならない。
2) 硫化物
廃水中の有機性浮遊物等が底泥土に沈降し、その分解によつて酸素が消費されて還元状態になると、硫酸塩還元細菌の増殖によつて硫化水素が発生し、これが底質中の金属等と硫化物を生成する。このため底質が悪変し、底棲生物の棲息に対して影響を与える。更に状態が悪くなると、底質から上層の水に対して二次的な汚染が起こる場合もある。
定量法はいろいろの方法があるが、ここでは試泥を水蒸気蒸留して硫化水素を追い出し、これを捕集してヨウ素滴定法で定量する方法を述べる。
蒸留する場合には、始め試料をそのまま蒸留して硫化物を測定し、その後酸を加えて更に蒸留し、硫化物を測定する。こうすると、浮遊硫化物はそのまま蒸留され、また酸性にして蒸留すると結合している硫化物も追いだされるので、厳密にではないが両者を分けて定量することが可能である。結果は過マンガン酸カリウム(0.1N)による酸素消費量と同じく乾燥試料1gあたりの硫化物イオウのmg数であらわす。
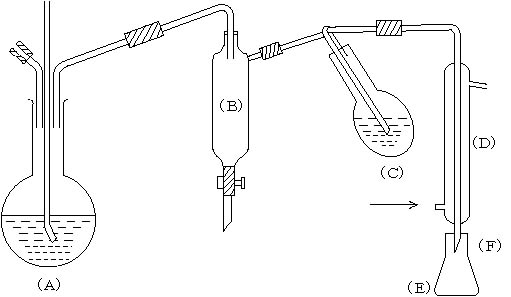
(1) 器具
水蒸気蒸留装置:水蒸気発生フラスコ(A)(容量1l丸底)円筒管(B)(容量500ml)・蒸留フラスコ(C)(容量300ml・丸底又はなす型)・冷却管(D)・受器(E)(容量100ml・三角フラスコ)よりなる(図―1)。冷却管(E)の先端には、先端を細長く引いたガラス管(F)をゴム管で接続する。窒素定量用のKjeldahl蒸留装置が用いうるのなら転用すれば便利である。ただし、混用はしない方がよい。もちろん冷却管の内管に銀管を用いてはならない。
(2) 試薬
ヨウ素溶液(0.01N):ヨウ素約1.3gをビーカーに取り、ヨウ化カリウム2g、水20mlを加えて溶解した後、全容を1lに希釈する。
チオ硫酸ナトリウム溶液(0.01N):チオ硫酸ナトリウム(Na2S2O3・5H2O)2.5gを水に溶解し1lとする。これに安定剤として、約0.1gの炭酸ナトリウムを加えておく。
(3) 試験操作
2―1)の分析試料20g(湿泥)をはかり取り、分解フラスコ(C)に移し、水20~30mlを加えて混和する。受器の三角フラスコ(E)中に酢酸亜鉛溶液(1W/V%)10mlを入れ、ガラス管注1)(F)の先端を受液中に浸す。水蒸気発生フラスコ(A)を加熱して水蒸気を通じ、蒸留を開始する。50~60ml受器中に留出したら、ガラス管(F)を冷却管からはずし、とりはずした管を入れたまま三角フラスコ(E)にヨウ素溶液(0.01N)5ml注2)、塩酸(1+1)2mlを加えてよくかき混ぜ、澱粉溶液を指示薬として0.01Nチオ硫酸ナトリウム溶液で滴定する。ここまでに遊離の硫化物が留出すると考えられる。次に、受器(E)とガラス管(F)を換え、分解フラスコ(C)中に塩酸(1+5)15mlを加え前操作と同じく蒸留し、留出液を滴定する。これで全硫化物が蒸留されたと考えることができる。
図―1 硫化物蒸留装置
試料の分析を行う前に、水を用いて空試験を行う必要がある。
〔結果の計算と表示〕
チオ硫酸ナトリウム溶液(0.01N)1mlは0.160mgの硫化物イオウに相当する。チオ硫酸ナトリウム溶液の滴定値を(t)ml、空試験値を(b)ml、その力価をfNa2S2O3・乾燥試料に対する重量百分率を(D)%とすれば、乾泥1gに対する硫化物は、次のように求められる。
硫化物(Smg/1g乾泥)=(0.16×{(d)-(t)}+fNa2S2O3×100/20×(D)
先述したように中性の蒸留において留出するものを遊離硫化物、酸性の蒸留で留出するものを結合硫化物、これらの和を全硫化物とし、すべてイオウのmg数であらわす。
- 注1) 蒸留の度ごとに取り換える方がよい。
- 注2) ヨウ素溶液(0.01N)5mlを加えても不足の場合は、過剰になるように5mlずつ追加する。この場合は空試験に用いたヨウ素溶液の容量すなわち5mlと、追加した時に加えたヨウ素溶液全量の比率を求め、空試験値にこの比率を乗じてこれをこの場合の空試験値とすればよい。
5 定量限界
|
項目
|
定量限界値(ppm)
|
|---|---|
|
総水銀
|
0.01
|
|
アルキル水銀
|
0.01
|
|
カドミウム
|
0.05
|
|
鉛
|
0.2
|
|
総クロム
|
1
|
|
6価クロム
|
2
|
|
ヒ素
|
0.5
|
|
BHC
|
0.01
|
|
PCB
|
0.01
|
この値は、通常の場合における定量限界値であり、現在環境庁が取りまとめに使用しているものである。
注) N、Dは定量限界値未満をいう。